
Abstract
Ewing sarcoma (ES) is a rare and highly-aggressive tumor. This malignancy derives from primitive neuroectodermal cells. The most frequently-affected population is adolescents and young adults, and the tumor typically grows in the bones. ES of the sinonasal tract is an exceedingly-rare situation. Particularly, primary occurrence in the ethmoid sinus is extremely scarce, with only 27 cases documented worldwide. Extension to the anterior cranial fossa is even thinner on the ground with limited cases in literature. We report the case of a 20-year-old female with history of Hodgkin disease presenting with facial pain, anosmia, and visual impairment on the right side. Imaging revealed a large invasive tumor originating from the right ethmoid sinus, with extension into the orbit and anterior cranial fossa. On biopsy, the tumor showed focal expression of Epithelial membrane antigen (EMA) and CD99 and EWSR1 rearrangement. The diagnosis of ES was confirmed. The patient underwent neoadjuvant chemotherapy with etoposide. Given the stability of the tumor, the patient was deemed inoperable. The decision was to proceed with chemoradiotherapy. Unfortunately, the patient passed away during treatment due to intracranial hypertension with seizures 12 months after diagnosis. This case underscores the rarity of sinonasal ES, particularly with primary involvement in the ethmoid sinus with anterior cranial fossa invasion. We conducted an exhaustive review of literature on similar cases to highlight the importance of a multidisciplinary approach combining advanced imaging, molecular diagnostics, chemotherapy, surgical intervention, and radiotherapy to optimize patients’ survival. Sinonasal ES, especially in the ethmoid sinus with cranial extension, is a rare and challenging situation. Detection at an early stage through meticulous imaging, immunohistochemistry, and molecular biology tools is crucial to initiate a multimodal treatment approach. Through this case report and literature review, we aimed to contribute to the growing literature on rare sinonasal malignancies.
Introduction
Ewing sarcoma (ES) is a rare small round cell malignancy belonging to the family of primitive neuroectodermal tumors. This class is subdivided into 2 categories. The first and central one is represented by neuroblastoma, whereas the peripheral category includes ES. 1 Due to its high metastatic potential, and little to no response to treatment, ES is classified as one of the most aggressive malignancies, mainly affecting adolescents and young adults. 2 In fact, it is ranked as the second most common primary malignancy of bones in subjects aged between 10 and 15 years old, after osteosarcoma. 3 Along with that, ES has the highest fatality rate. 4 The most common location is the chest and the pelvis. 5 Extraskeletal involvement is a rare situation, and location in the head and neck accounts for <4% of all ES locations. Sinonasal tract involvement is an exceedingly-rare situation, and primary ES of the ethmoid sinus is a scarce and challenging situation with only few cases reported thus far. 6 We aimed to report a rare case of primary ES of the ethmoid sinus extending into the orbit and the anterior cranial fossa. As we delve into its misleading clinical presentation, its particular imaging features, as well as the diagnostic challenges overcame through immunohistochemistry and molecular biology, we aimed to highlight the importance of a multidisciplinary approach in facing the fatality of such rare and complex cases. By reporting this case alongside a review of the literature, we aimed to provide insights into the sparse existing literature on this subject.
Case Report
A 20-year-old female patient was referred to our center with persistent unilateral sinonasal symptoms. Medical history revealed stage IIB Hodgkin lymphoma, treated successfully a year and a half ago with 2 cycles of ABVD Adriamycin, Bleomycin, Vinblastine, Dacarbazine) and BEACOPP (Bleomycin, etoposide, Adriamycin, cyclophosphamide, Procarbazine, Prednisone), followed by radiotherapy, achieving complete remission. Eighteen months later, the patient was presented with facial pain, right-sided proptosis, nasal obstruction, and anosmia, which had been evolving over 3 months. On examination, she was presented with a WHO performance status of 1, right-sided proptosis with restricted ocular movement and orbital swelling. Nasal endoscopy revealed a mass occupying the right nasal cavity.
Computed tomography (CT) scan revealed an irregular infiltrative tissue mass centered on the right ethmoid, measuring 60 mm in its largest axis, extending posteriorly to the sphenoidal sinus, laterally to the medial walls of the orbits, creating an intraorbital fatty mass on the right, causing homolateral exophthalmos (Figure 1A). The right extraocular muscle cone was thinned, with the disappearance of the fatty planes, extending to the root of the nose. The mass extended into the nasal fossae, predominantly on the right, with stenosis of the ostium and complete obliteration of the maxillary sinus. From the right ethmoidal sinus, the tumor extended through the cribriform plate (Figure 1B). The right cribriform plate was lysed, thus creating a right-sided cerebral mass measuring 55 mm in the anterior cranial fossa.
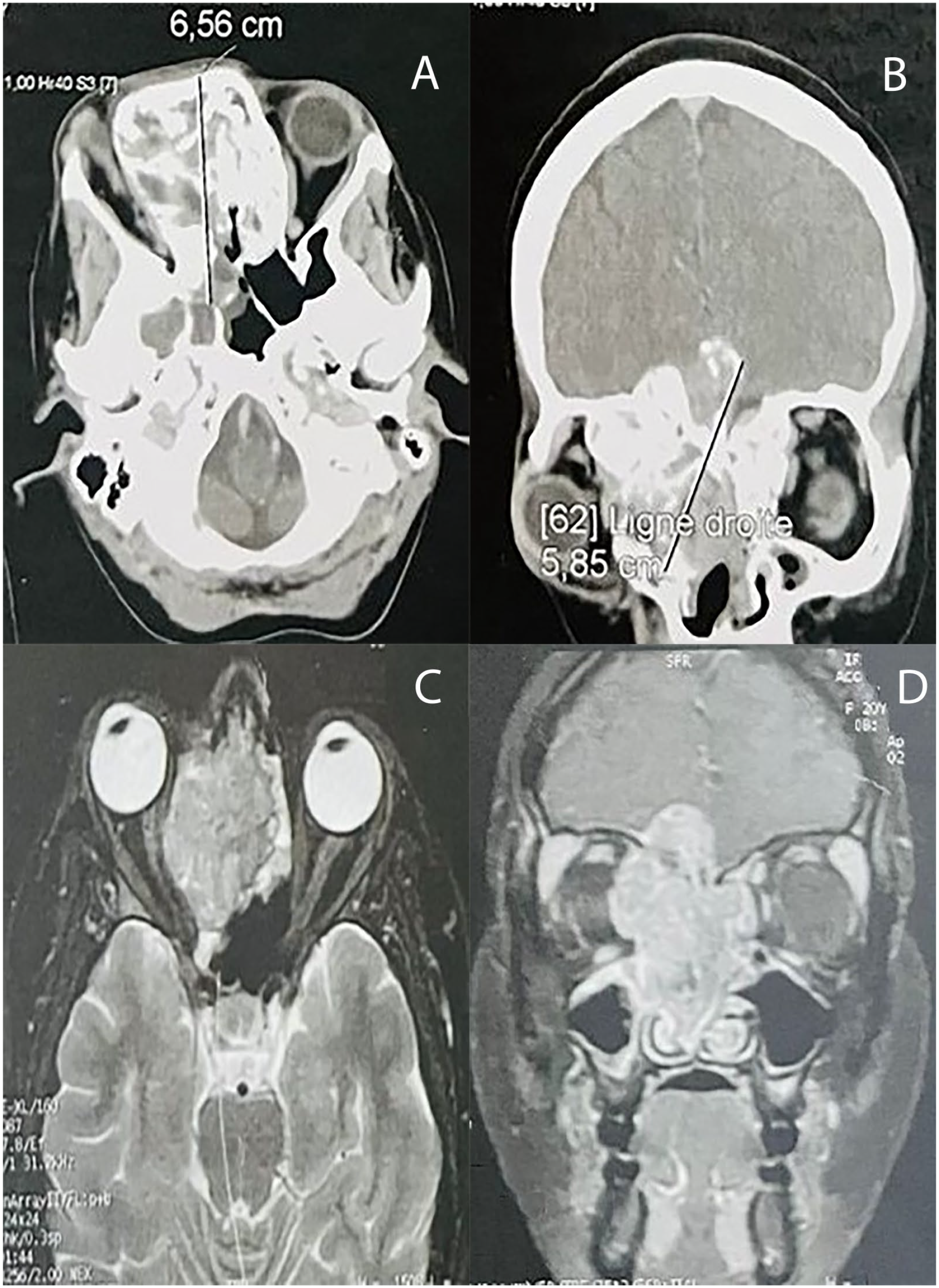
(A) Axial facial CT scan showing irregular infiltrative tissue mass centered on the right ethmoid, measuring 60 mm in its largest axis, extending posteriorly to the sphenoidal sinus, laterally to the medial walls of the orbits, creating an intraorbital fatty mass on the right, causing homolateral exophthalmos. (B) Coronal facial CT scan showing intracranial extension of the tumor through the cribriform plate. (C) Axial T2-weighted MRI revealing the ethmoidal lesion displacing the medial wall of the right orbit, compressing the medial rectus muscle and the optic nerve, and causing grade I exophthalmos. (D) Injected T1-weighted coronal MRI revealing endocranial extension into the right anterior cranial fossa, crossing the cribriform plate and pushing the right olfactory bulb upward. CT, computed tomography; MRI, magnetic resonance imaging.
Magnetic resonance imaging (MRI) revealed a tumoral tissue process centered on the right ethmoid, measuring 6 × 3 × 5 cm. The lesion occupied almost the entirety of the right nasal fossa, displacing the medial wall of the right orbit, compressing the medial rectus muscle and the optic nerve, and causing grade I exophthalmos (Figure 1C). The superior and middle turbinates were lysed by the lesion. It displaced the nasal septum into the left ethmoidal hemi-space and extended to the level of the right choana. The lesion showed an endocranial extension into the right anterior cranial fossa, crossing the cribriform plate and pushing the right olfactory bulb upward (Figure 1D).
No secondary cerebral lesions were detected. Retention of fluid was noted in the maxillary sinuses, and the cavum mucosal reliefs were intact. The parapharyngeal fat had a normal appearance.
A prior biopsy realized outside of our center revealed angiofibroma of the nasal fossa. Another biopsy in our center suggested inflammatory changes without malignancy. A third biopsy was performed and showed a small round blue cell tumor (Figure 2A), moderately atypical, with focal necrosis (Figure 2B), and highly vascularized. The mitotic index was high and reached 30 mitoses/10 high-power fields. Periodic acid-Schiff (PAS) staining showed granular cytoplasmic positivity (Figure 2C). A large panel of immunohistochemistry including epithelial markers (CK, p63, p40) (Figure 2D), melanin markers (PS100, Melan A), neuroendocrine markers (chromogranin, synaptophysin, CD56), striated muscle markers (desmin, myogenin, sarcomeric actin), and vascular markers (CD34) was negative.
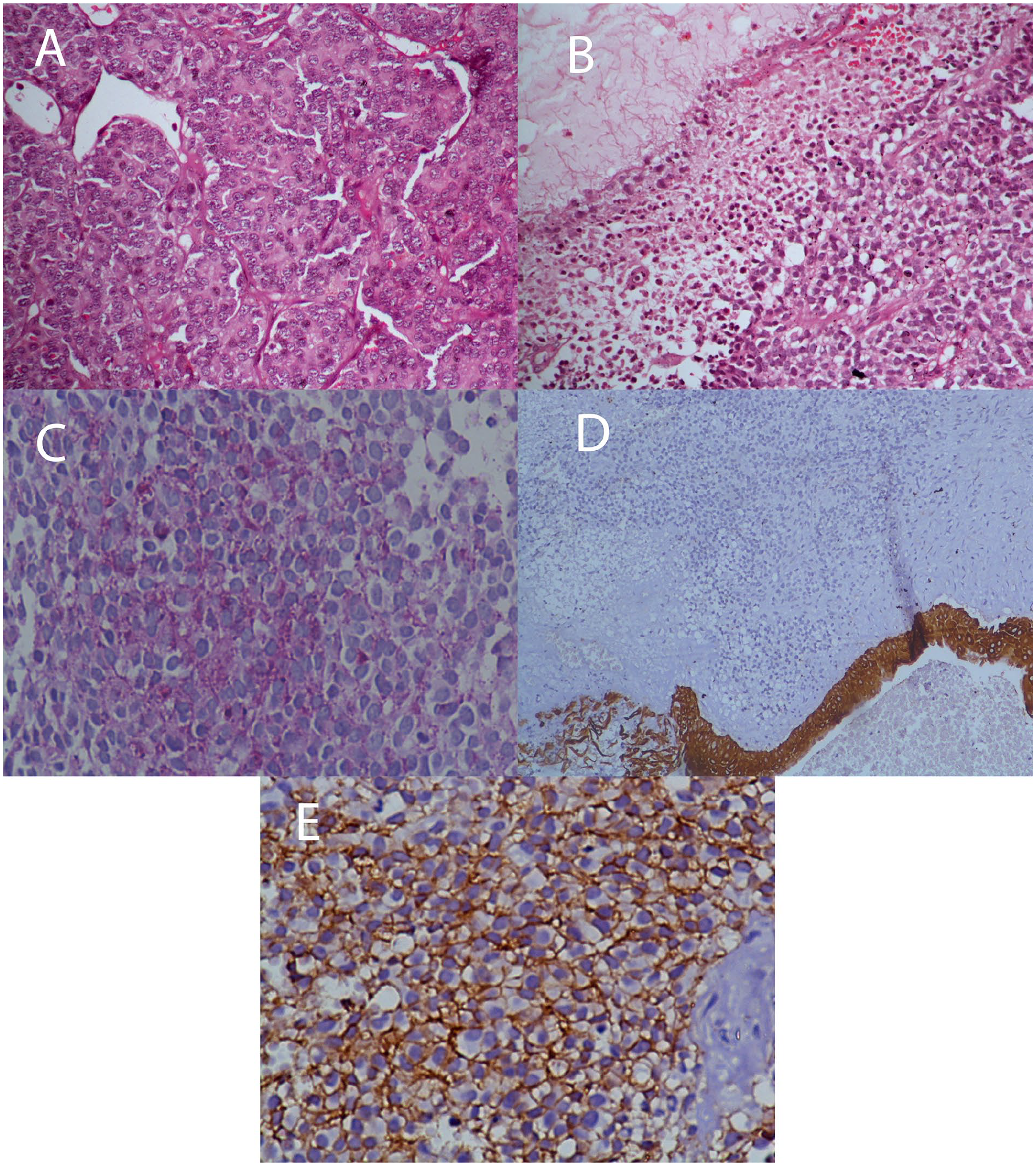
(A) Ewing sarcoma composed of solid areas of uniform undifferentiated round cells with indistinct cell borders and scattered mitotic cells (arrows; Hematoxylin-Eosin (HE) ×400). (B) Section revealing necrosis (HE ×200). (C) PAS showed granular cytoplasmic positive staining (PAS ×400). (D) CK showed negative staining with positive internal control at the bottom (CK ×100). (E) CD99 showed strong, membranous staining in the tumor cells (CD99 ×400).
The tumor showed only focal but strong expression of EMA and CD99 (Figure 2E) and EWSR1 rearrangement.
Bone marrow biopsy showed no evidence of tumor infiltration. PET-CT revealed a hypermetabolic lesion confined to the ethmoid sinus, without distant metastases. The patient was considered inoperable due to the massive orbital and endocranial extension. A multidisciplinary decision was made to administer chemotherapy first and then reassess the extension. Prior to therapy, fertility preservation was assured with ovocyte cryopreservation. She received 5 cycles of VIP chemotherapy (etoposide, ifosfamide, cisplatin). A subsequent CT scan revealed stability in size and extension of the calcified ethmoidal tumor (Figure 3).
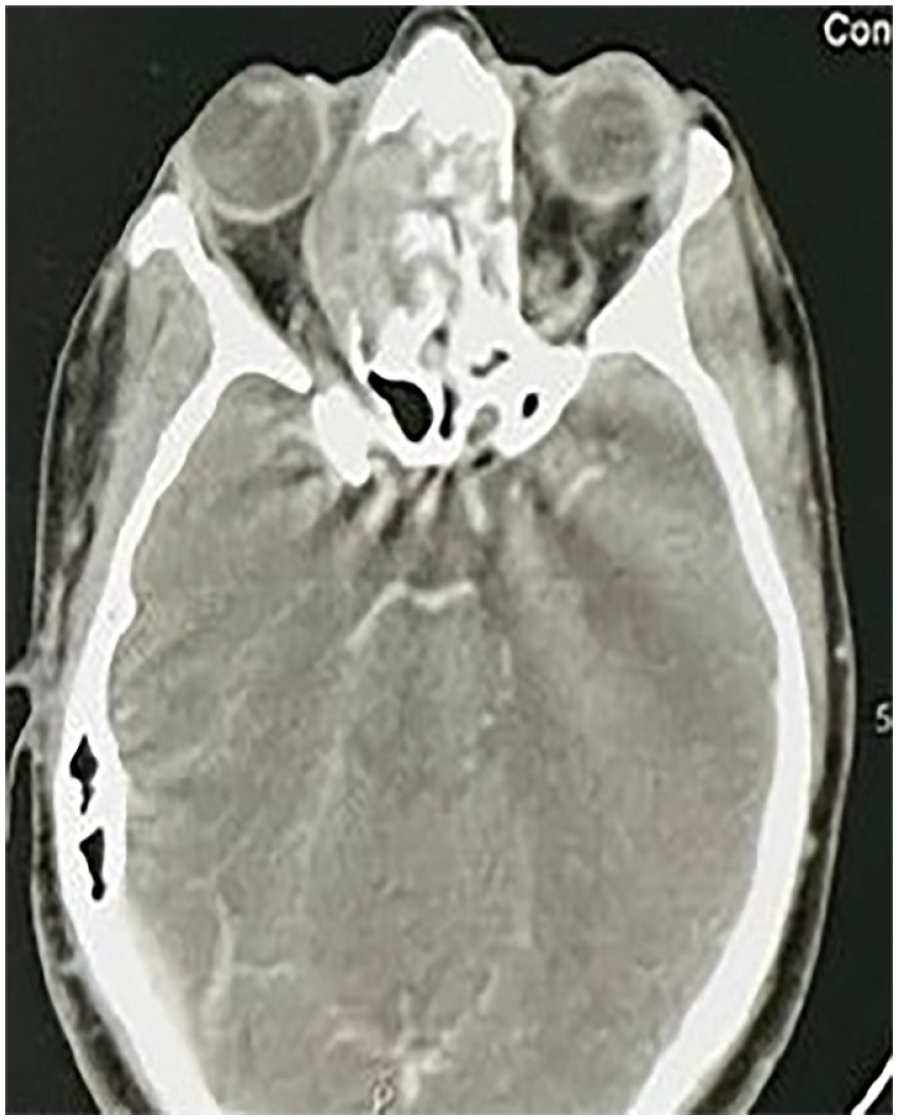
Subsequent computed tomography scan revealing stability in size and extension of the calcified ethmoidal tumor.
The decision as made to proceed with radiotherapy in conjunction with chemotherapy based on etoposide cisplatin. She received a dose of 22 Gy with only 2 cycles of chemotherapy. The patient experienced significant toxicities, grade 4 mucositis, hearing loss, and persistent vomiting. Unfortunately, she passed away in the context of intracranial hypertension with seizures 12 months after diagnosis.
Discussion
Described by the american pathologist James Ewing for the first time in 1921, ES is a rare aggressive malignancy predominantly arising in the skeleton of adolescents and young adults. 1 The most typical sites are the long bones and the pelvis where the tumor originates from the neuroectodermal cells of the medullary cavity. 7 Extraskeletal ES involving soft tissues without bone involvement, accounts for a smaller subset of cases. Among the rarer sites, the head and neck region constitutes <4% of ES cases, with the paranasal sinuses being particularly uncommon. 8 Within the sinonasal tract, the primary involvement of the ethmoid sinus is exceedingly rare. 9 Given the rarity of this location, this tumor is often misdiagnosed as other sinonasal malignancies, such as sinonasal undifferentiated carcinoma, neuroendocrine carcinoma, rhabdomyosarcoma, or lymphoma. 10
A thorough literature review was carried out, revealing a very limited number of cases of primary ethmoid ES (Table 1).
Exhaustive Review of Literature on Reported Cases of Primary Ewing Sarcoma of the Ethmoid Sinus.
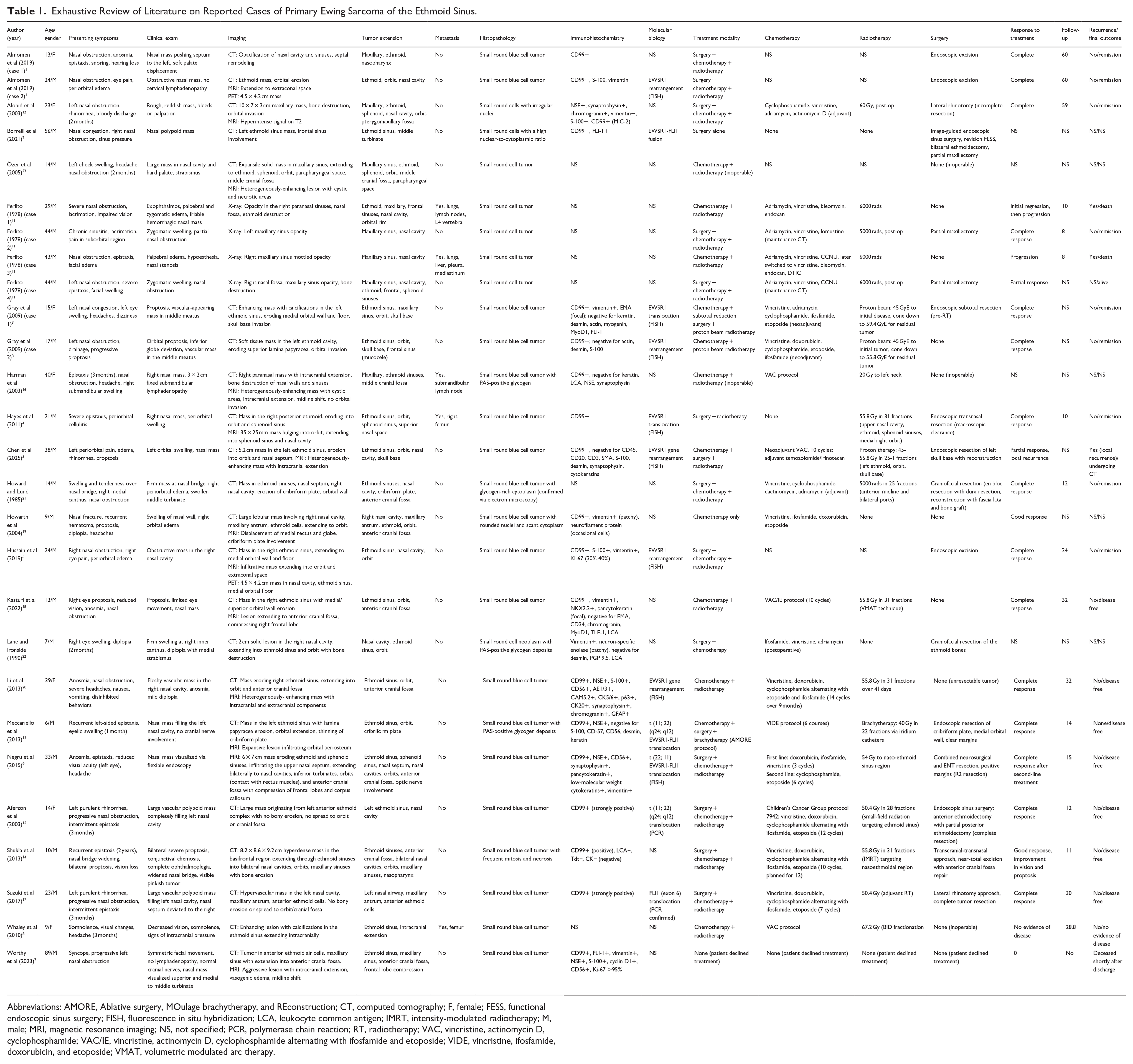
Abbreviations: AMORE, Ablative surgery, MOulage brachytherapy, and REconstruction; CT, computed tomography; F, female; FESS, functional endoscopic sinus surgery; FISH, fluorescence in situ hybridization; LCA, leukocyte common antigen; IMRT, intensity-modulated radiotherapy; M, male; MRI, magnetic resonance imaging; NS, not specified; PCR, polymerase chain reaction; RT, radiotherapy; VAC, vincristine, actinomycin D, cyclophosphamide; VAC/IE, vincristine, actinomycin D, cyclophosphamide alternating with ifosfamide and etoposide; VIDE, vincristine, ifosfamide, doxorubicin, and etoposide; VMAT, volumetric modulated arc therapy.
The first ES of the sinonasal tract was reported by Ferlito in 1978 who documented the case of a 44-year-old male with an ES of the ethmoid extending on X-ray to the right nasal fossa and maxillary sinus opacity with. The case was successfully managed with surgery followed by chemoradiotherapy. 11 Since then, a small number of cases have been documented, shedding light on the demographic, clinical, and pathological aspects of this entity. The average age at diagnosis of these cases is approximately 23 years. The age limits span from 6 to 89 years.6,7 More than half of the cases were male patients, accounting for 60% of cases.8,9
The most frequent symptoms were nasal obstruction and epistaxis, reported in over 80% and 50% of cases, respectively.5,12 Ophthalmologic symptoms such as proptosis and diplopia were observed in locally-advanced cases where the tumor extends to the orbit.3,13 Less frequent symptoms include anosmia, headaches, or cranial nerve deficits.14,15 Metastases were reported in almost 20% of cases and were documented either at the time of diagnosis or during follow-up. The most common metastatic sites were the lungs and the bones (such as the femur and vertebrae).8,16
To establish a thorough evaluation of such tumors, imaging tools play a crucial role. On CT, these lesions typically display as enhancing soft tissue masses with bony destruction, involving the ethmoid sinus walls, lamina papyracea, and cribriform plate.17,18 However, MRI allows assessing soft tissue extension as it reveals heterogeneous contrast enhancement with cystic or necrotic areas. Furthermore, MRI is superior when it comes to delineating intracranial extension and orbital invasion.9,19 The most common extension sites are the orbit and anterior cranial fossa.1,2 Less frequent sites of extension was documented in the sphenoid sinus, pterygomaxillary fossa, and nasopharynx.4,20
As for pathology examination, it routinely reveals small round blue cells with scant cytoplasm and a high nuclear-to-cytoplasmic ratio. These cells classically show high mitotic activity with areas of necrosis.21,22 Immunohistochemical analysis is crucial to establishing diagnosis. In fact, a strong membranous positivity of CD99 is a nearly-universal characteristic across cases.6,14 Other frequently expresses markers by ES include vimentin, FLI-1, NSE, and Synaptophysin. On the other hand, negative staining for desmin, cytokeratin, and leukocyte common antigen is critical to rule out differential diagnoses such as rhabdomyosarcoma, carcinoma, and lymphoma.9,20 Molecular biology is key in confirming the presence of EWSR1 gene rearrangements. Particularly, the EWSR1-FLI1 fusion is identified via fluorescence in situ hybridization or reverse transcription-polymerase chain reaction.5,15
When it comes to treating sinonasal ES, the protocols are classically multimodal. Options combine chemotherapy, radiotherapy, and surgery if it is feasible. Chemotherapy frequently involves VAC/IE protocols (vincristine, actinomycin D, cyclophosphamide alternating with ifosfamide and etoposide). This can be administered either as neoadjuvant or adjuvant therapy.3,23 Radiotherapy is often employed with doses ranging from 50.4 to 67.2 Gy. The radiation is delivered through advanced techniques such as intensity-modulated radiotherapy or proton beam therapy, particularly in cases with intracranial or orbital extension.7,19 Surgical resection is done if it is anatomically feasible, depending on orbital and cerebral extension. Endoscopic transnasal approaches are preferred for localized tumors. However, craniofacial resections are reserved for locally-advanced cases with skull base extension.9,13
A particular treatment plan was recently published, the AMORE (ablative surgery, moulage brachytherapy, and reconstruction) protocol. This regimen aims to enhance local tumor control simultaneously minimizing long-term treatment-related morbidity. This protocol involves neoadjuvant chemotherapy followed by complete surgical resection. Interventions are often carried out via an endoscopic approach. This protocol is combined with mold-based brachytherapy using iridium catheters and associated with subsequent reconstruction. The AMORE protocol has proved oncological success. Furthermore, it reduced functional and aesthetic complications in such young populations. 13
In most cases, therapeutic outcomes have been favorable. with complete responses achieved through aggressive multidisciplinary protocols.1,15 However recurrences are possible, particularly in cases with intracranial extension or incomplete surgical resection.8,14 Mortality was also reported among these rare cases. One of them involved a 29-year-old male who developed multiple metastases to the lungs and vertebrae despite chemoradiotherapy. This disease progression led to death within 10 months. 11 Another documented death was a 17-year-old male with ethmoid ES extending to the brain from initial presentation. This patient experienced local recurrence and distant metastasis despite high-dose radiotherapy and chemotherapy. The patient ultimately dies of disease after 27 months. 8 Additionally, another case documented an 89-year-old male with extensive intracranial disease who declined treatment. Death was shortly documented after hospital discharge. 7
Conclusions
Primary ES originating in the ethmoid sinus is a rare and aggressive malignancy, particularly with extension to adjacent organs, like the orbit and the brain. Despite its challenging diagnosis, with imaging and clinical features resembling other sinonasal malignancies, it requires a precise and prompt diagnosis. A critical tool to achieving this is molecular biology as it confirms EWSR1 translocation. A multidisciplinary approach is key to achieving oncological control. This multimodal management, involving chemotherapy, surgical resection with skull base reconstruction, and radiotherapy, has proved its success in many cases.