
Abstract
Ewing’s sarcoma (EWS) is a rare malignant bone tumor that primarily affects children and young adults. While it typically affects long bones, it can occur in the head and neck region, including the paranasal sinuses in rare cases. We present a challenging case of a 45-year-old female diagnosed with an EWS of the sphenoidal and the right nasal fossa. A subtotal removal of the mass was performed on the patient followed by combined adjuvant radiotherapy and chemotherapy. Due to its rarity, the diagnosis and management of EWS in the paranasal sinuses are challenging.
Introduction
Ewing’s sarcoma (EWS) is an aggressively malignant tumor arising from primitive neuroectodermal cells.1-3 This rare malignant bone tumor typically affects long bones in children and young adults. 1 It ranks as the second most common malignancy affecting bones and soft tissues in childhood. 4 EWS of the head and neck region is extremely uncommon, representing only 4% to 9% of all EWSs. It typically affects the mandible and calvaria. 3 Additionally, sinonasal lesions are infrequent. 1
We present a case of a patient with a primary sarcoma originating in the sphenoidal sinus. Our objective is to discuss the clinical characteristics of primary Ewing sarcomas of the sinus tract and their management modalities.
Case Report
A 45-year-old female with a previous history of papillary thyroid cancer stage pT1b N1b M1 underwent comprehensive treatment, including total thyroidectomy, bilateral central neck dissection, bilateral functional cervical lymph node dissection, radiation therapy, and adjuvant radiotherapy, along with metastectomy of the 8th rib, presented with a 5-month history of right nasal obstruction, chronic rhinorrhea, cacosmia, and intermittent epistaxis.
An endoscopic examination revealed an irregular, mamillated tissue formation with a friable component exhibiting a pearly tint. This mass completely fills the right nasal fossa, invades the body of the lower turbinate, and protrudes backward into the nasopharynx with a suspected infiltration of its lateral wall (Figure 1).
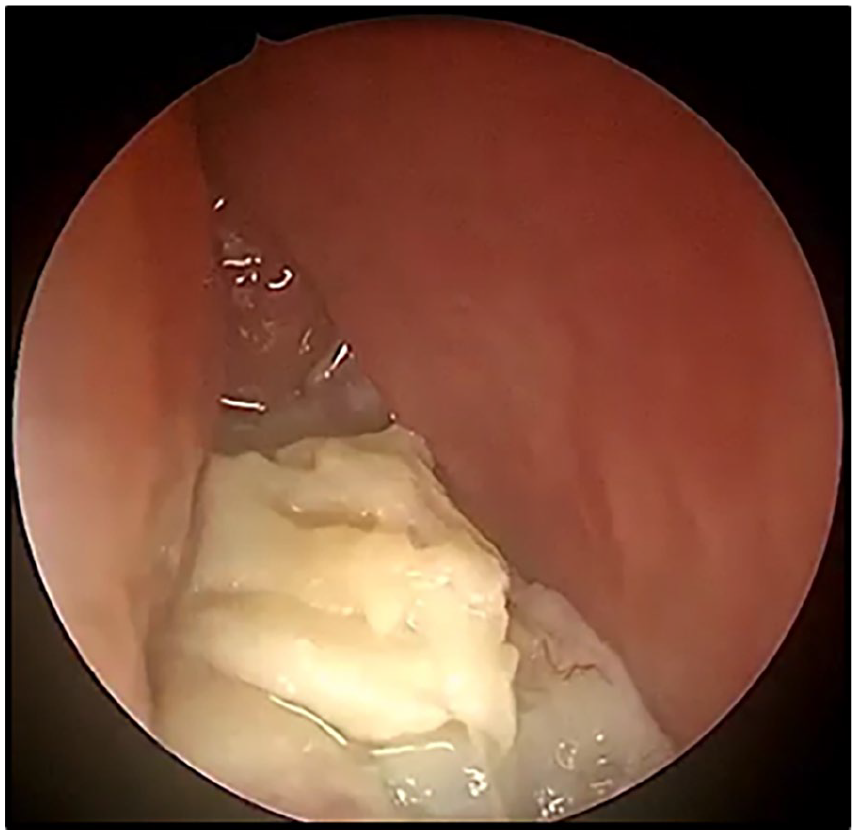
Endoscopic examination showed a large irregular polypoid mass with a friable pearly white component that completely fills the right nasal fossa.
Computed tomography (CT) revealed a heterogeneous tissue mass affecting the right choanae, the sphenoidal sinus, the right ethmoidal cells, and the nasopharynx. The contours of this lesion are lobulated. There is no evidence of invasion into the skull base (Figure 2).
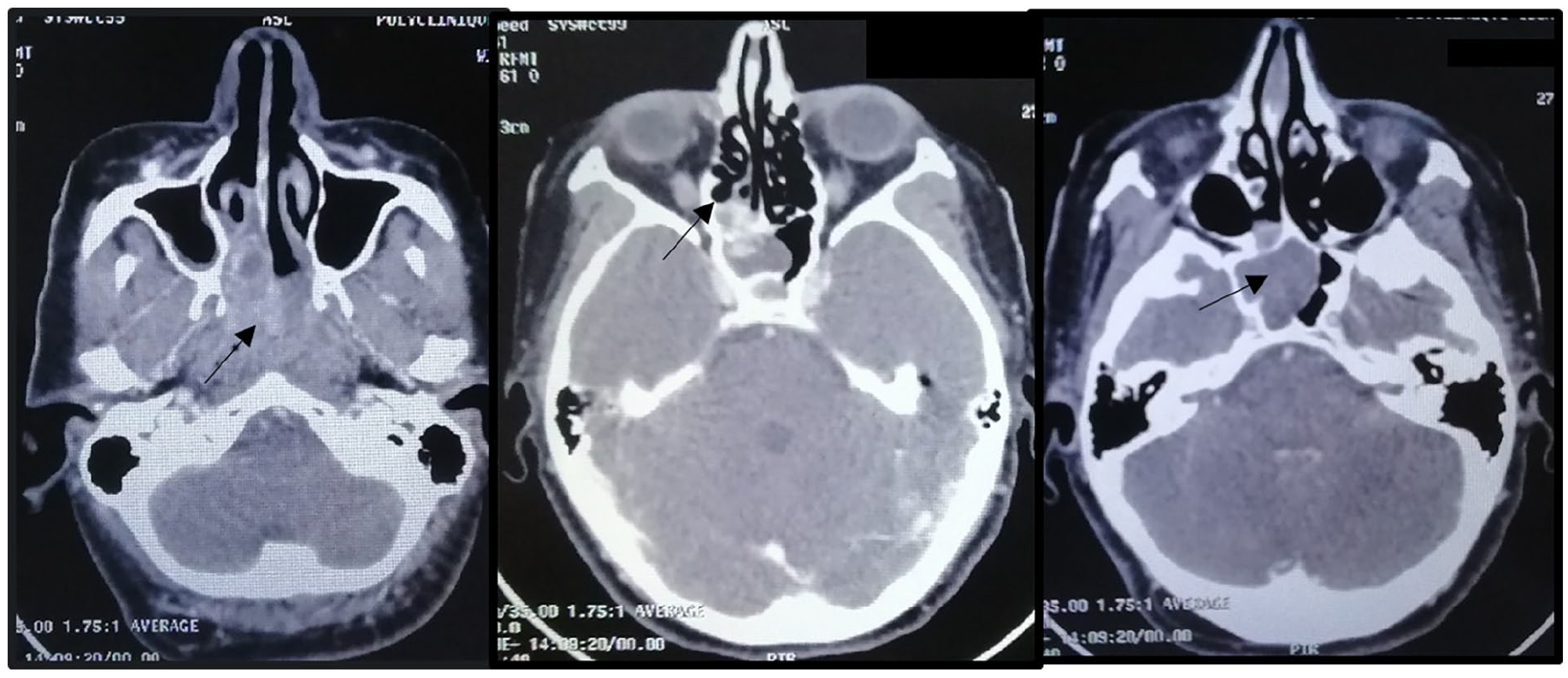
CT scan showed a large heterogeneous enhancing mass involving the right posterior ethmoid cells, sphenoidal sinus, and nasopharynx. CT, computed tomography.
Magnetic resonance imaging (MRI) showed a large lobulated mass involving the right posterior ethmoid cells, sphenoidal sinus, nasopharynx, and reaching the oropharynx. This lesion exhibits T2 hypointensity and T1 isointensity. After gadolinium injection, the tumor showed a heterogeneous enhancing mass with a predominant peripheral pattern. There was no extrasinus extension, involvement of the skull base, orbit, and bone erosion in the MRI (Figure 3).
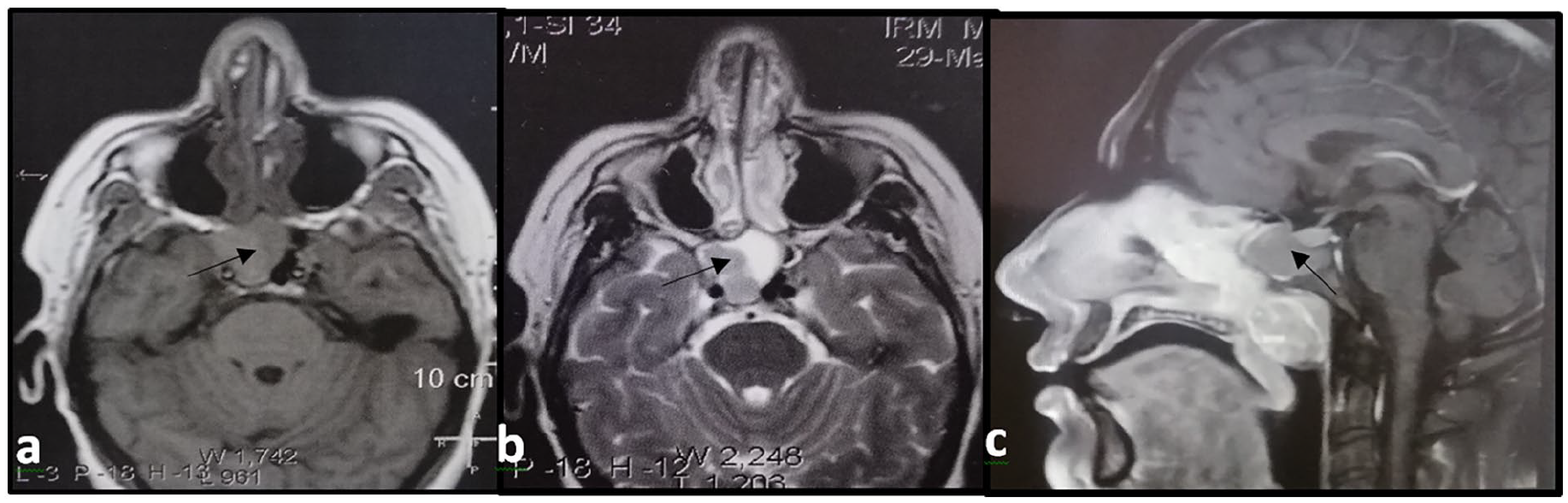
MRI axial (a, b) and sagittal (c) views showing a lesion of the ethmoid and sphénoidal sinus, bilateral nasal cavities, and nasopharynx, with T1 isointensity (a) and T2 hypointensity (b). MRI, magnetic resonance imaging.
The patient underwent a total removal of the mass with right anterior and posterior ethmoidectomy, and sphenoidotomy, along with biopsy of the nasopharynx.
The histological and immunohistochemical examination revealed Ewing sarcoma of the sphenoidal and right nasal fossa. The mucosa of the ethmoid, maxillary sinus, and nasopharynx appear normal, without tumor infiltration.
Microscopic examination revealed malignant tissue with a proliferative growth often necrotic, occasionally compressed, arranged in diffuse sheets, and sometimes in lobules separated by a vascular network. The tumor cells are relatively monomorphic, round, and of small to medium size, with indistinct cytoplasmic borders. The cytoplasm is scant, sometimes eosinophilic, and sometimes cleared. Nuclei are round, with fine chromatin and occasional small nucleoli. Mitoses are numerous. The stroma is moderately abundant- and fi+bro-inflammatory. The tumor is ulcerated, occasionally bordered by granulation tissue (Figure 4).
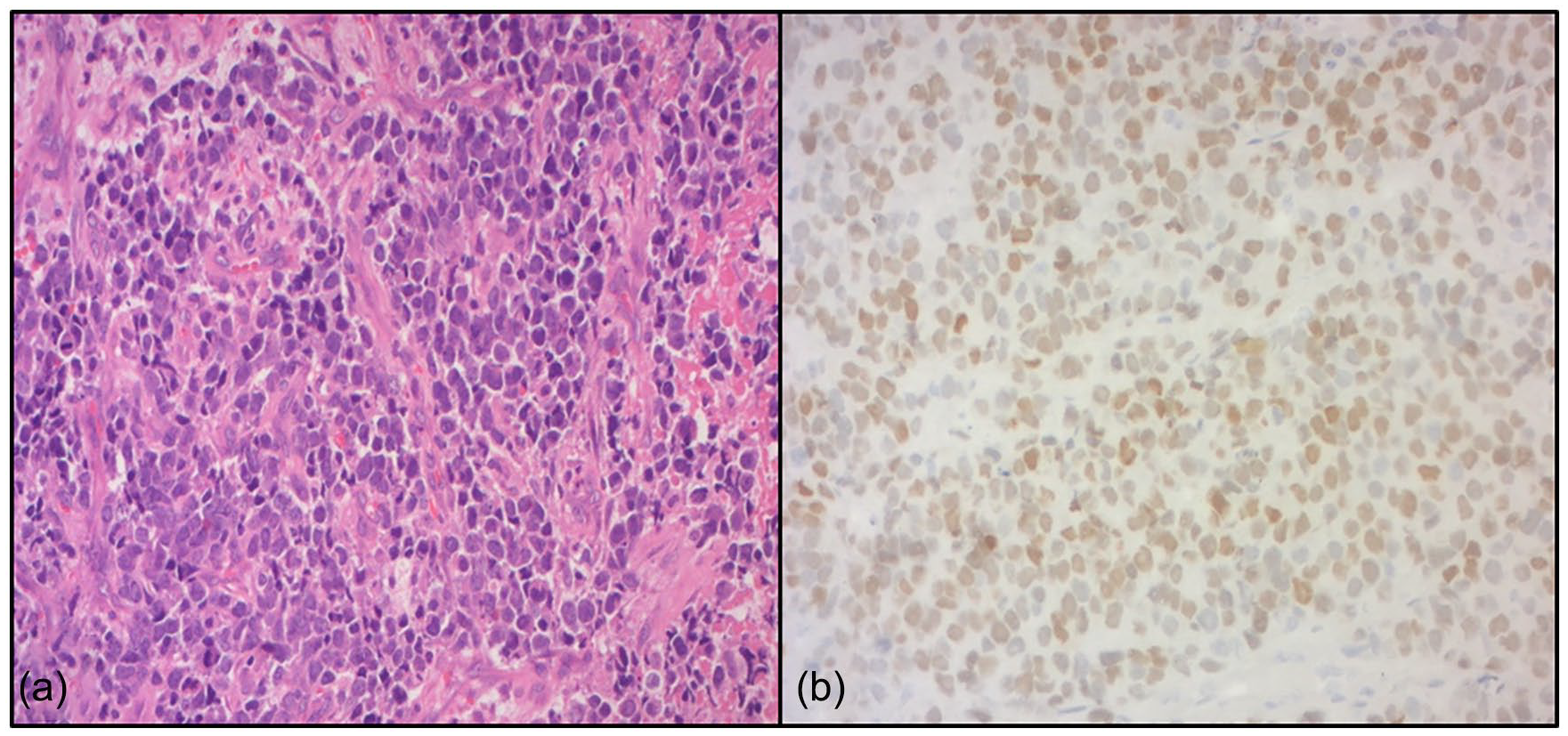
Microscopic analysis. (a) Undifferentiated malignant tumor with round cell (HE ×200). (b) Diffuse ant intense nuclear immunohistochemical positivity for NKX2.2 (HE ×200).
Immunohistochemical examination revealed positive staining for CD99, INI-1, and NKX 2-2, and negative staining for keratin, desmin, calretinin, PS100, and chromogranin. Focal positivity was observed for p40 and synaptophysin.
Postoperatively, the patient was treated with combined radiotherapy and chemotherapy of Vincristine, Ifosfamide, Doxorubicin, and Etoposide.
A 3 months follow-up MRI showed no recurrence. After 2 years of follow-up, the patient did not have tumor recurrence.
Discussion
EWS is an extremely malignant tumor originating from primitive neuroectodermal cells. 1 The initial description of this tumor was provided by James Ewing in 1921. 1 EWS most often develops in early childhood, reaching its peak around the age of 15 years.1,2 It is infrequently observed in adulthood. 1 It is likely to be more common in men. 1 EWS in the head and neck region is exceptionally rare, accounting for only 4% to 9% of all EWS cases. 1 The most frequent location for primary head and neck EWS is the temporal bone, followed by the frontal, parietal, and occipital bones. 2 Sphenoid and ethmoid bones are less frequently affected. 2
No specific environmental factors have been associated with EWS. 4 However, treating primary cancer in childhood, especially with radiation therapy, appears to elevate the risk of developing EWS. 4
The clinical presentation of EWS of sinus tract is variable and is affected by the location and the size of the tumor. 4 The most common symptom is headache and pain with a rapidly enlarging mass.2,3
CT and MRI play an important role in the diagnosis of EWS and provide important information for surgical treatment planning. CT is the reference imaging, providing information on cortical bone expansion, erosion, and subsequent destruction. 3 MRI evaluates the extension of the lesion into adjacent soft tissues. 3
The differential diagnosis of EWS is tumors with small round cells, including olfactory neuroblastoma, rhabdomyosarcoma, lymphoma, melanoma, and undifferentiated carcinoma.1-3
On microscope examination, these tumors are composed of uniform small round cells characterized by circular nuclei containing fine chromatin, along with minimal clear or eosinophilic cytoplasm. 1 The diagnosis of EWS, includes a histopathological examination, immunohistochemical testing, and cytogenetic analysis. 1 The CD99 marker is essential to establish the diagnosis. In our case, the patient tested positive for CD99 marker.
EWS is also characterized by the translocation of a gene, situated on 22q12, with the FLI1 gene located on 11q24, resulting in a t(22;11) translocation, which gives rise to an EWS–FLI1 fusion gene observed in 85% of EWS cases. Detection of these fusion genes can be achieved through either fluorescence in situ hybridization or reverse transcriptase polymerase chain reaction (RT-PCR).1,3,5
The treatment of EWS is a multidisciplinary approach. The baseline treatment involves neoadjuvant chemotherapy followed by a surgical resection and radiotherapy. Currently, there is still no general consensus about the management of these tumors.
Neoadjuvant chemotherapy (4–6 cycles) is recommended even when the tumor is resectable as it eradicates micro-metastatic disease and improves the local control by ensuring extensive negative margins. 3
The surgical procedure should be radical and in instances of a positive marginal resection, it will be associated with radiotherapy. Adjuvant chemotherapy is recommended in the standard approach. It is based on a combination of Doxorubicin, Cyclophosphamide, Vincristine, Actinomycin D, Ifosfamide, and Etoposide. 5
The combination of surgery, radiation, and chemotherapy enhanced loco-regional control of the disease, leading to a notable improvement in the 5-year survival rate, increasing from 10% to 50%–65%.5,6
In general, EWS of the head and neck tends to have a favorable prognosis when contrasted with EWS occurring in other locations. 7 Prognosis varies with many factors such as patient’s age, comorbidities, tumor location, and the presence of metastasis.3,8
Patients diagnosed with metastasis exhibit a considerably worse prognosis compared to those without metastasis with a long-term survival rate of only 20% for individuals with metastasis. 5
Conclusion
It is uncommon for EWSs to affect the paranasal sinuses or nasal fossa. A multidisciplinary approach, combining surgical resection and/or radiotherapy with chemotherapy, has the potential to improve long-term survival.
Footnotes
Acknowledgements
All authors approved the final version and have the agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethical approval for reporting individual cases.
Patient Consent
Written informed consent obtained from the patient.