
Abstract
A 71-year-old man with neurofibromatosis type 1 (NF1) presented to our department with a 1-week history of a painful mass in the left submandibular area. Computed tomography (CT) and magnetic resonance imaging revealed an irregular-shaped tumor with a diameter of 2.0 cm in the left submandibular gland and a metastatic lymph node with a diameter of 1.0 cm adjacent to the tumor. Fluorodeoxyglucose–positron emission tomography/CT revealed increased uptake in the tumor. Fine-needle aspiration cytology revealed atypical cells, suggesting salivary duct carcinoma (SDC). Left neck dissection with resection of the tumor and submandibular gland was performed under general anesthesia. Histologic examination revealed ductal formation with a solid, cystic, cribriform, and papillary structure with intraductal comedonecrosis, diagnosing as SDC originating in the submandibular gland (pT3N1M0 pStage III). Mutational analysis of 160 cancer-related genes by next-generation sequencing (NGS) revealed a germline and frameshift mutation in the NF1 gene (p.R2408Kfs*14) and a somatic and frameshift mutation in the TP53 gene (p.C176Wfs*22). The patient received postoperative radiotherapy to the left neck area at 66 Gy. No evidence of recurrence or metastasis has been observed as of 10 months postoperatively. This is the first reported case of SDC in the submandibular gland in a patient with NF1. The mutational data by NGS may contribute to a better understanding of the oncogenesis of SDC in patients with NF1.
Keywords
Introduction
Salivary duct carcinoma (SDC) is an uncommon tumor originating in the salivary gland and histologically resembles high-grade breast ductal carcinoma. 1 SDC is regarded as a high-grade malignant tumor, exhibiting clinically aggressive behavior with locoregional recurrence and distant metastasis. The molecular mechanism of the carcinogenesis of SDC remains unknown. Neurofibromatosis type 1 (NF1), also known as von Recklinghausen’s disease, is an autosomal dominant inherited disorder caused by mutation in the NF1 gene and impacts approximately 1 in 3000 births. 2 The common signs and symptoms include café-au-lait macules, neurofibromas, skinfold freckling, Lisch nodules, and optic pathway gliomas. 3 Patients with NF1 often develop malignant tumors such as gliomas and malignant peripheral nerve sheath tumor (MPNST). Breast carcinoma often occurs in patients with NF1; however, SDC in patients with NF1 has not been reported. This article describes a rare case of SDC arising in the submandibular gland in a patient with NF1 and the results of mutational analysis using next-generation sequencing (NGS).
Case Presentation
A 71-year-old man with NF1 presented to our department with a 1-week history of a painful mass in the left submandibular area. His mother and older sister were diagnosed with NF1. A thumb-sized hard nodule was palpated on the left submandibular area and neurofibromas covered not only the neck but also the entire surface of his body with café-au-lait macules (Figure 1A). Contrast-enhanced computed tomography (CT) scan revealed a slightly enhanced irregular-shaped tumor with a diameter of 2.0 cm in the left submandibular gland and a metastatic lymph node with a diameter of 1.0 cm adjacent to the tumor (Figure 1B). T1-weighted gadolinium-enhanced magnetic resonance imaging (MRI) revealed that the tumor had heterogeneous enhancement (Figure 1C). Fluorodeoxyglucose (FDG)–positron emission tomography (PET)/CT scan revealed increased uptake in the tumor of the left submandibular gland and a metastatic lymph node (Figure 1D). Fine-needle aspiration cytology (FNAC) demonstrated small- to large-sized clusters of atypical cells with a high nucleus/cell ratio, oval to pleomorphic nuclei, and large and prominent nucleoli, suggesting SDC (Figure 2A).
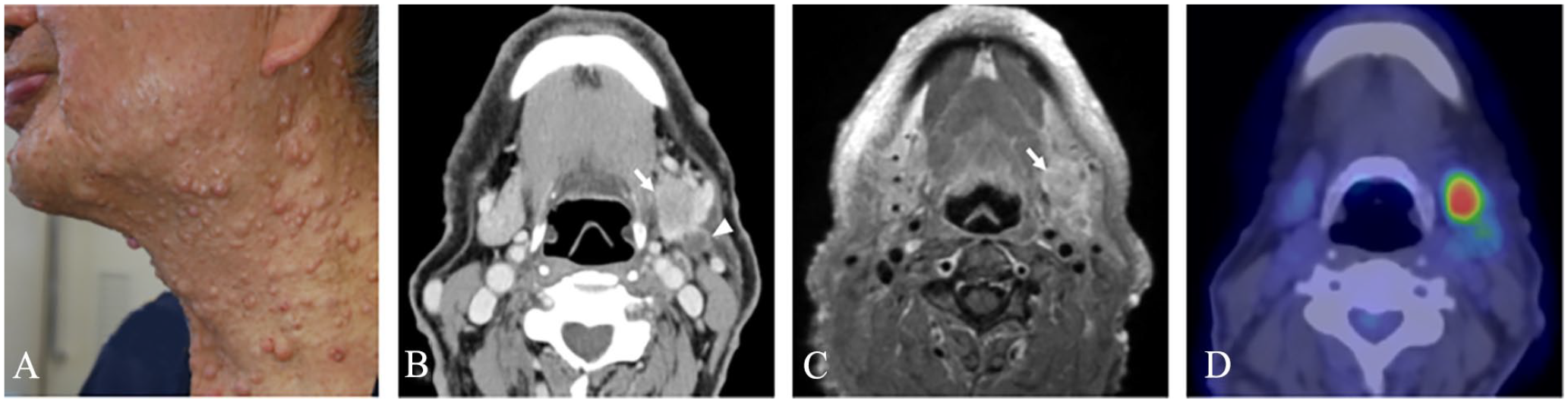
Imaging findings of the tumor. A thumb-sized hard nodule was palpated on the left submandibular area and the neck was covered with neurofibromas (A). Contrast-enhanced CT scan revealed a slightly enhanced irregular-shaped tumor with a diameter of 2.0 cm in the left submandibular gland (arrow) and a metastatic lymph node with a diameter of 1.0 cm adjacent to the tumor (triangle) (B). Gadolinium-enhanced MRI on T1-weighted images revealed that the tumor had heterogeneous enhancement (arrow) (C). FDG-PET/CT revealed increased uptake in the tumor of the left submandibular gland and a metastatic lymph node (D).
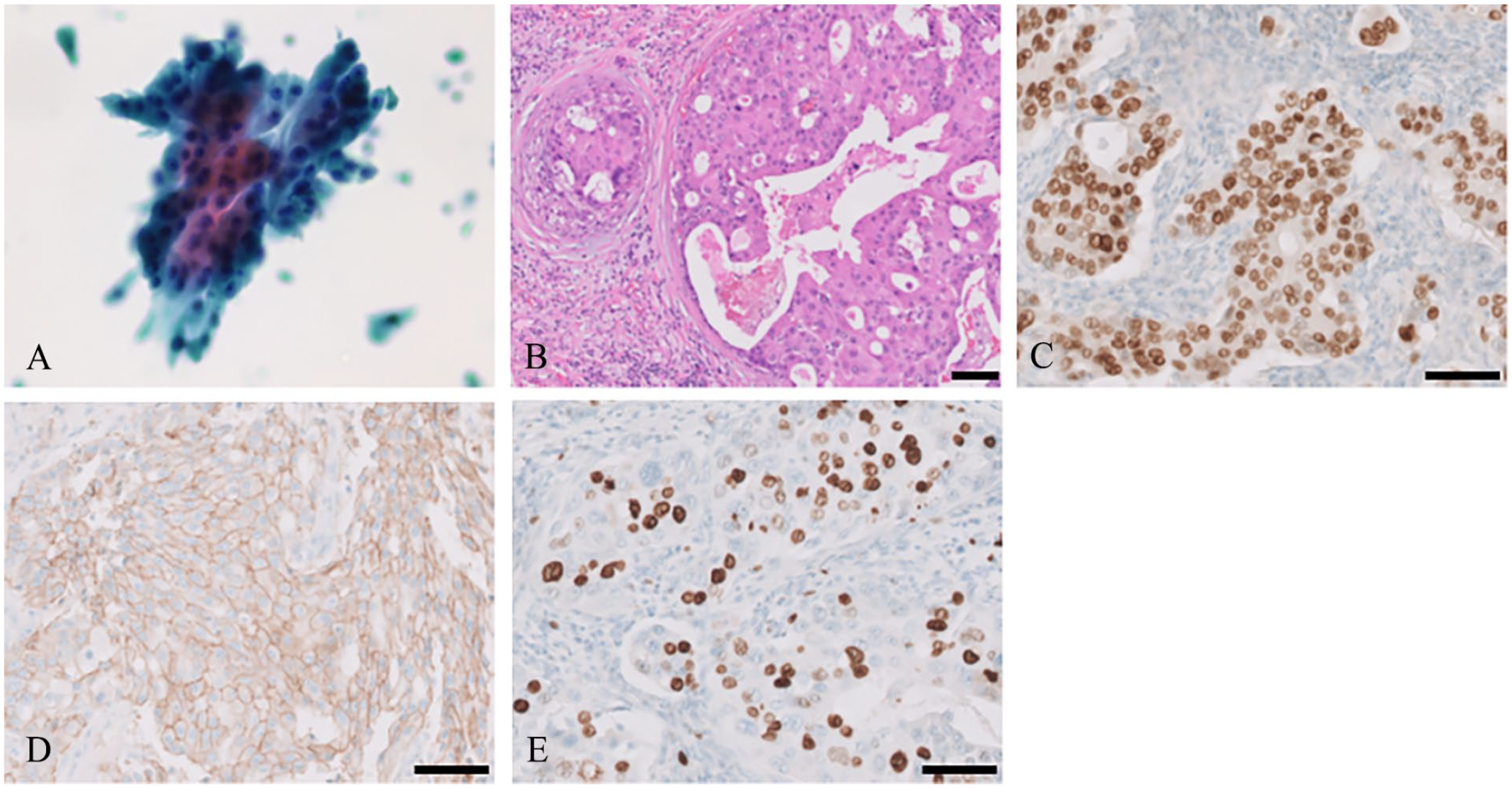
Cytologic and histologic findings of the tumor. FNAC demonstrated small- to large-sized clusters of atypical cells with high nucleus/cell ratio, oval to pleomorphic nuclei, and large and prominent nucleoli, suggesting SDC (A). Histologic examination revealed ductal formation with a solid, cystic, cribriform, and papillary structure with intraductal comedonecrosis (B). Immunohistologic examination revealed positivity for AR (1+) (C) and HER2 (2+) (D). The percentage of Ki-67-positive cells was approximately 70% (E). All scale bars: 100 μm.
Left neck dissection from level I to V with resection of the tumor and the submandibular gland was performed under general anesthesia. The skin incision was made through an arcuate incision from the submental area to the mastoid process with a longitudinal incision at the midclavicular line. The left submandibular ganglion and the lingual nerve were resected due to invasion by the tumor. Histologic examination revealed ductal formation with a solid, cystic, cribriform, and papillary structure with intraductal comedonecrosis (Figure 2B). Immunohistologic examination revealed positivity for the androgen receptor (AR; 1+, Figure 2C) and human epidermal growth factor receptor 2 (HER2; 2+, Figure 2D). The percentage of Ki-67-positive cells was approximately 70% (Figure 2E). The final diagnosis was made as SDC in the submandibular gland (pT3N1M0 pStage III).
NGS panel testing of targeted regions of 160 cancer-related genes was performed with tumor samples and peripheral blood molecular cells as previously described (Supplemental Table S1),4,5 revealing a germline and frameshift mutation in exon 49 of the NF1 gene (c.7219_7220insC; p.R2408Kfs*14) (Figure 3A) and a somatic and frameshift mutation in exon 5 of the TP53 gene (c.525_556delCTGCCCCCACCATGAGCGCTGCTCAGATAGCG; p.C176Wfs*22) (Figure 3B). No other pathologic mutations were found in the cancer-related genes. The patient received postoperative radiotherapy to the left neck area at 66 Gy/33Fr. No evidence of recurrence or metastasis has been observed as of 10 months postoperatively.
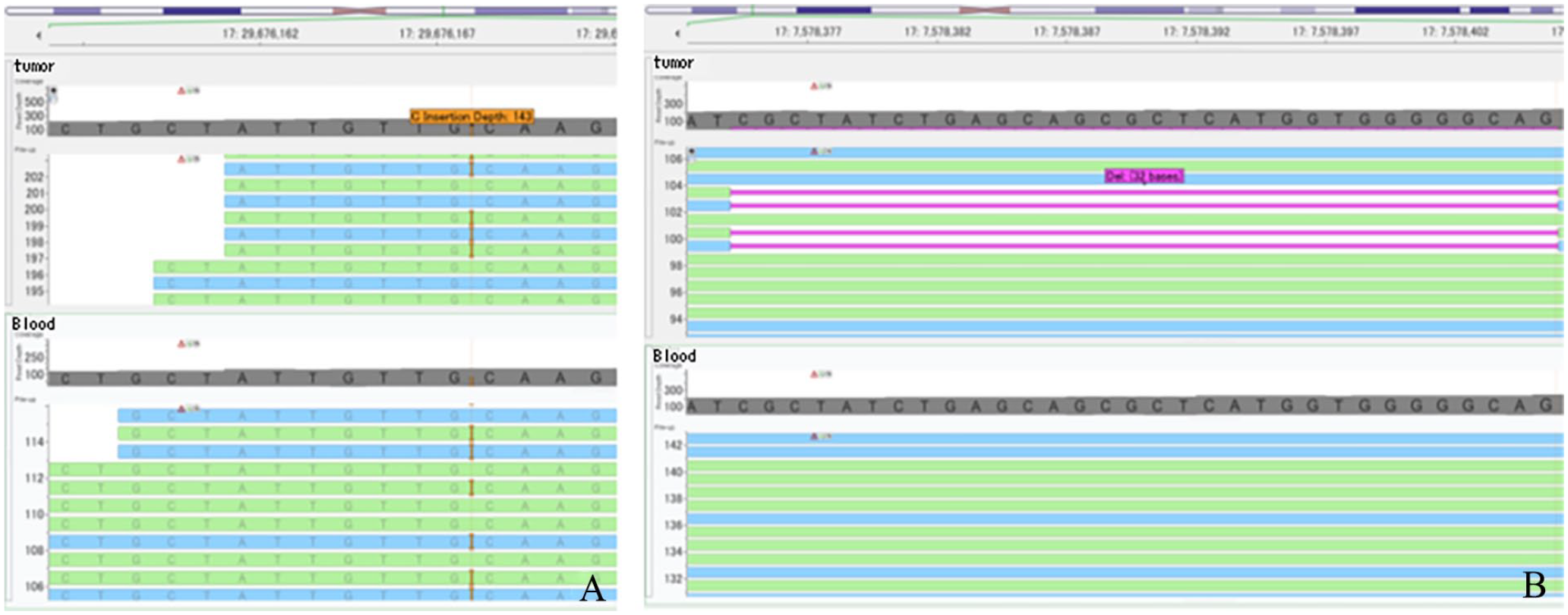
Mutational analysis by NGS. Targeted genomic DNA sequences determined were compared between tumor samples and peripheral blood molecular cells. A germline and frameshift mutation in exon 49 of the NF1 gene (c.7219_7220insC; p.R2408Kfs*14) (A) and a somatic and frameshift mutation in exon 5 of the TP53 gene (c.525_556delCTGCCCCCACCATGAGCGCTGCTCAGATAGCG; p.C176Wfs*22) (B) were detected. Images were produced using the free software Golden Helix Genome Browse (http://goldenhelix.com) and then modified.
Discussion
SDC accounts for approximately 10% of salivary gland carcinomas. In a study of 141 SDC patients, the tumor most commonly originated in the parotid (79.4% of cases), followed by the submandibular gland (17.7% of cases) or other locations (3% of cases). 6 SDC reportedly develops at approximately 60-70 years of age, with a male-to-female incidence ratio of approximately 3:1. 7 FNAC is an accurate and reliable method for classifying SDC as a malignant neoplasm but it is much less accurate for identifying the specific tumor type. 8 SDC is diagnosed based on histologic characteristics, including the presence of a complex solid, cribriform, and papillary-cystic architecture with frequent comedonecrosis. Tumor cells have large pleomorphic nuclei with coarse chromatin and prominent nucleoli, along with abundant eosinophilic, typically apocrine, cytoplasm. 9 In one study, tumor cells were positive for AR and HER2 in 71.9% and 43.8% of 32 patients, respectively. 1 Resection with an appropriate safety margin of the tumor with neck dissection followed by radiotherapy is the standard treatment for SDC of the submandibular gland with neck lymph node metastasis. 6 The prognosis of SDC is reportedly unfavorable, with a 5-year survival rate of only 23.2% due to local recurrence and/or metastasis. 10 Chemotherapy with trastuzumab and docetaxel is used in patients with HER2-positive, recurrent, or metastatic SDC. 11 To date, there have been no reports of SDC arising in the submandibular gland in patients with NF1.
NF1 results from a germline mutation in the NF1 gene on chromosome 17q11.2. 12 Any mutation in one germline allele is sufficient to result in the syndrome of NF1. In the present case, the patient with NF1 had a germline and frameshift mutation in the NF1 gene (p.R2408Kfs*14). To date, more than 500 mutations in the NF1 gene have been identified. 13 The types of germline mutations in a study examining 160 patients with NF1 were nonsense in 36.6%, missense in 24.4%, splicing site mutations in 14.6%, frameshifts in 12.2%, microdeletions in 8.5%, and whole gene deletions in 3.8%. 14 Neurofibromin, which is encoded by the NF1 gene, has a central region with a marked structural and sequence similarity to ras-guanosine-triphosphate (GTP)ase activation proteins (GAPs). GAPs inactivate Ras by accelerating the conversion of active Ras-GTP to its inactive guanosine diphosphate-bound form.13,15 Functional loss of neurofibromin due to inactivating mutations or deletions in the NF1 gene leads to aberrant over-activation of the RAS signaling cascade and increased downstream activity of oncogenic mitogen-activated protein kinase and phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin signaling pathways. 16 In addition, the tumor suppression system, including the promotion of apoptosis, regulation of cell adhesion and motility, and suppression of heat-shock proteins and epithelial–mesenchymal transition, is reportedly inhibited by the functional loss of neurofibromin. 17
Patients with NF1 tend to develop malignant tumors, primarily in the central and peripheral nervous systems. Life expectancy in patients with NF1 is reportedly 10-15 years lower than in those without NF1. 18 In patients with NF1, the overall risk of malignancy is increased by approximately 2.7 times, and more malignant tumors tend to develop at a younger age compared to those without NF1. 18 Therefore, the prognosis of patients with NF1 depends on whether malignant tumors develop and treatment outcomes. 19 A large-scale study of 1607 patients with NF1 reported the development of various malignant tumors, including low-grade gliomas in 16.6%, MPNST in 15.1%, breast carcinoma in 2.9%, high-grade glioma in 1.7%, pheochromocytoma in 1.2%, gastrointestinal stromal tumors in 1.2%, melanoma in 0.9%, and thyroid carcinoma in 0.4%. 19 The report indicated that patients with NF1 develop not only tumors in the nervous system but also various malignant tumors, including carcinomas.
In the present case, a somatic and frameshift mutation in the TP53 gene (p.C176Wfs*22) was identified by NGS panel testing using tumor samples from SDC in the submandibular gland. The TP53 gene contributes to tumor suppression through two mechanisms in response to DNA damage: arrest of cell proliferation and induction of apoptosis. 20 A study of 125 patients with SDC in the salivary glands reported that apart from NF1, TP53 mutations were the most frequent genetic mutation in 68% of patients, followed by mutations in PIK3CA in 18% and H-RAS in 16%. 21 The types of mutations in the TP53 gene were reportedly missense in 61%, nonsense in 9.4%, and frameshift mutations in 27% of cases. Patients with SDC and TP53 mutations showed shorter progression-free survival. 21
Monoallelic loss of NF1 is likely to increase the potential for coordination with other pathways, such as the TP53 pathway, to promote cellular proliferation and carcinogenesis. 22 Any germline mutation in the NF1 gene can potentially affect other genes on chromosome 17, which include the gene encoding tumor suppressor protein p53 at 17p13.2, HER2 at 17q21.1, and breast cancer gene 1 at 17q21.2. 17 In another study, TP53 mutations were identified in 17.5% of 40 patients with malignant tumors and NF1 germline mutations. 23 The coexistence of germline NF1 and somatic TP53 mutations has also been observed in sporadic tumors such as malignant astrocytoma, 24 melanoma, 25 and breast carcinoma 17 in patients with NF1. The TP53 gene was mutated in 73% of 41 patients with ovarian carcinoma with documented NF1 mutation, suggesting that the pathways regulated by these two tumor-suppressor proteins often cooperate in the development of ovarian carcinomas with serous differentiation. 26 Another study identified TP53 mutations in 80% of patients with breast carcinomas along with NF1 and suggested that coordinated effects of the NF1 and TP53 mutations can trigger carcinogenesis, resulting in the development of an aggressive phenotype. 27 Results of these reports suggested that coordinated effects of NF1 and TP53 mutations in the present study might be associated with carcinogenesis of SDC.
Clinically, the diagnosis of breast carcinoma in patients with NF1 is often delayed due to the presence of cutaneous fibromatosis in the trunk. 28 This may apply to neck tumors, including salivary gland carcinomas and metastasis to neck lymph nodes, due to the presence of cutaneous fibromatosis of the head and neck. Radiation therapy was performed after surgical treatment in the present case considering the high-grade malignancy and invasive behavior of the SDC. Radiation therapy is known to cause secondary malignant tumors such as MPNST, and the risk is particularly elevated in children with NF1. 29 Therefore, for the follow-up in the present case, imaging studies such as CT scanning of the neck and lung and MRI of the brain are essential for the detection of locoregional recurrence, metastasis, radiation-induced secondary malignant tumors, or brain tumors, which often appear in patients with NF1.
Conclusions
We report a rare case of SDC in the submandibular gland in a patient with NF1. NGS panel testing revealed a germline mutation in the NF1 gene and a somatic mutation in the TP53 gene. Coordinated effects of both mutations might be associated with the carcinogenesis of SDC. In patients with NF1, clinicians should be aware of the possibility of salivary gland carcinoma.
Supplemental Material
sj-xlsx-1-ear-10.1177_01455613241231146 – Supplemental material for Salivary Duct Carcinoma Arising in the Submandibular Gland in a Patient with Neurofibromatosis Type 1
Supplemental material, sj-xlsx-1-ear-10.1177_01455613241231146 for Salivary Duct Carcinoma Arising in the Submandibular Gland in a Patient with Neurofibromatosis Type 1 by Shuto Hayashi, Nobuyuki Bandoh, Misaki Hayashi, Takashi Goto, Yasutaka Kato, Shogo Baba, Eriko Aimono and Hiroshi Nishihara in Ear, Nose & Throat Journal
Footnotes
Authors’ Note
This material has not been published elsewhere, either in whole or in part. All authors have been personally and actively involved in the substantive work of preparing the manuscript and are jointly and individually responsible for its content.
Authors’ Contributions
SH, NB, MH, and TG conducted the surgery and provided bedside care. SH and NB drafted the manuscript. YK and SB performed mutational analyses. EA and HN performed pathologic investigations. All authors approved the final version of the manuscript.
Data Availability Statements
The data that support the findings of this study are available from the corresponding author, NB, upon reasonable request.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
All procedures performed on patient tumor samples in this study were carried out in accordance with the ethical standards of the Institute Ethics Committee and the Declaration of Helsinki of 1964 and its subsequent amendments or comparable ethical standards. The present study was approved by the Ethics Committee of Hokuto Hospital (approval no. 1078; Obihiro, Japan).
Informed Consent
Written informed consent was obtained from the patient and his family for the publication of clinical details and images.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.