
Abstract
Neurofibromatosis type 1 (NF1) is a neurocutaneous condition with an autosomal dominant pattern of inheritance. This congenital disease is characterized by a wide spectrum of clinical manifestations and degree of severity. This case report describes a female patient in her early 20s who presented with a complaint of lumbosciatica-like pain evolving for several months. The condition initially escaped the attention of clinicians until a lumbar computed tomography scan and spinal magnetic resonance imaging were performed. The patient was then transferred to the general surgery department, where a clinical diagnosis of NF1 was established. The clinical manifestations were specific for this disease, including café-au-lait macules, plexiform neurofibroma, and a history of neurofibromatosis in her mother. The patient underwent surgical resection of the neurofibroma, which resulted in a favorable outcome. However, 2 years later, a new mass attached to the second lumbar spinal nerve was revealed by a follow-up computed tomography scan. Long-term and close follow-up of NF1 is required because of the high risk of malignancy and recurrence in NF1 patients.
Keywords
Introduction
Neurofibromatosis type 1 (NF1) (#MIM 162200), which is commonly referred to as Recklinghausen’s disease or peripheral neurofibromatosis, is the most common type of neurofibromatosis, which comprises a wide variety of hereditary tumor-predisposing syndromes characterized by the development of multiple tumors spread around the body. 1 The incidence rate of NF1 is estimated to be one in every 2500 to 3000 births, and patients with NF1 have an approximately 8% to 13% higher risk of developing malignant sarcomas than the general population (0.001%). Furthermore, the condition shortens the life expectancy to 54 years. 2
NF1 results from mutations in the NF1 gene, which encodes neurofibromin and is located on the 17q11.2 locus. 3 Neurofibromin is a GTPase-activating protein that serves as a negative regulator of the RAS/mitogen activated protein kinase (MAPK) signaling and mechanistic target of rapamycin (mTOR) pathways. 4 NF1 has an autosomal dominant pattern of inheritance, and its phenotypic manifestations may vary widely.5,6 Given this pattern of inheritance, the risk of mutation occurrence in an offspring is 50%, and in familial genetic counseling, this value is used to establish a diagnosis. 7
The diagnosis is established clinically based on the National Institutes of Health consensus developed in 1987 and includes at least two of the following criteria: coffee-colored patches (>5 mm diameter in prepubertal individuals and >15 mm in postpubertal individuals) throughout the body (macules), axillary or inguinal freckles, optic glioma, at least two neurofibromas of any type or one plexiform neurofibroma, and a distinctive osseous lesion (sphenoid dysplasia or tibial pseudarthrosis). 2 However, other clinical manifestations not included in this list may also occur and can affect the cardiovascular, gastrointestinal, renal, and endocrine systems. 8 One study reported facial and body dysmorphism and cognitive deficits, which highlight the importance of other diagnostic modalities, such as familial genetic counseling. 8 Here, we report a case of fortuitous discovery of a para-rectal neurofibroma in a female patient in her early 20s in whom NF1 was also newly clinically diagnosed. This case further confirms that surgical resection of neurofibromas can be achieved safely and constitutes a reminder for the clinical manifestations of this pathological entity for physicians.
Case report
A female patient in her early 20 s sought consultation for lumbosciatica-like pain that had evolved for several months. The interview revealed that she had a family history of NF1 diagnosed in her mother, who underwent surgery for NF1 of the small intestines.
The physical examination showed a good general condition, dorso-lumbar scoliosis, and skin lesions of different sizes (café-au-lait spots) (Figure 1). The neurological examination did not reveal any sensory or motor disorders, and the abdominal examination showed no anomalies. On the basis of the interview, NF1 was proposed as a diagnosis. This diagnosis had escaped the attention of many clinicians, and this was the first time that the patient was diagnosed with NF1. Biological tests did not reveal any anomaly. A lumbar computed tomography (CT) scan and spinal magnetic resonance imaging (Figure 2) revealed a right, latero-rectal, pre-sacral pelvic tissue mass that was located opposite to the right piriformis muscle and adjacent to the branches of the sciatic nerve. This mass was encapsulated, had a regular contour with T1 hyposignal and heterogeneous T2 hypersignal, had no diffusion restriction, and had heterogeneous enhancement that was more marked at the late stage. It measured 41 × 48 mm in the axial plane and 50 mm in height. The imaging results did not reveal any other associated adenomegaly. The remaining pelvic organs had no abnormalities. The patient underwent a colonoscopy that revealed a regular, oval, 50-mm-long lesion located 8 cm from the anal margin (Figure 3) stressing the mucosa of the middle rectum.
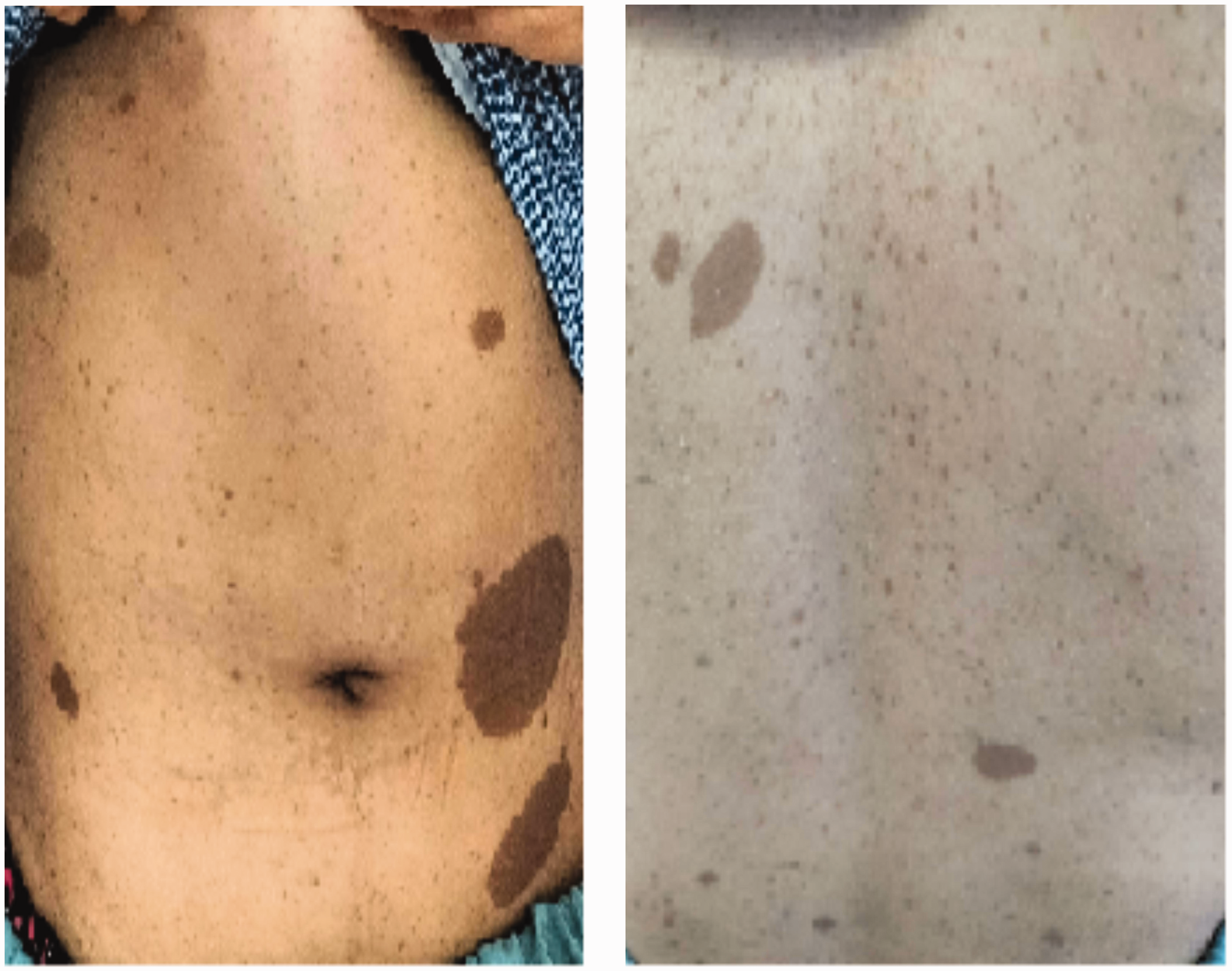
Cutaneous lesions (café-au-lait macules).

Spinal cord magnetic resonance imaging highlighting lesion aspects (X).

Coloscopy outcome: lesion exerting a mass effect on the rectal wall, bulging into the rectal lumen.
The patient underwent surgery via a pararectal incision, and we performed a complete exeresis of the lesion (Figure 4). The intervention proceeded without incident and postoperative sequalae as revealed by a normal neurological examination. The pathological examination indicated that the mass was a neurofibroma.
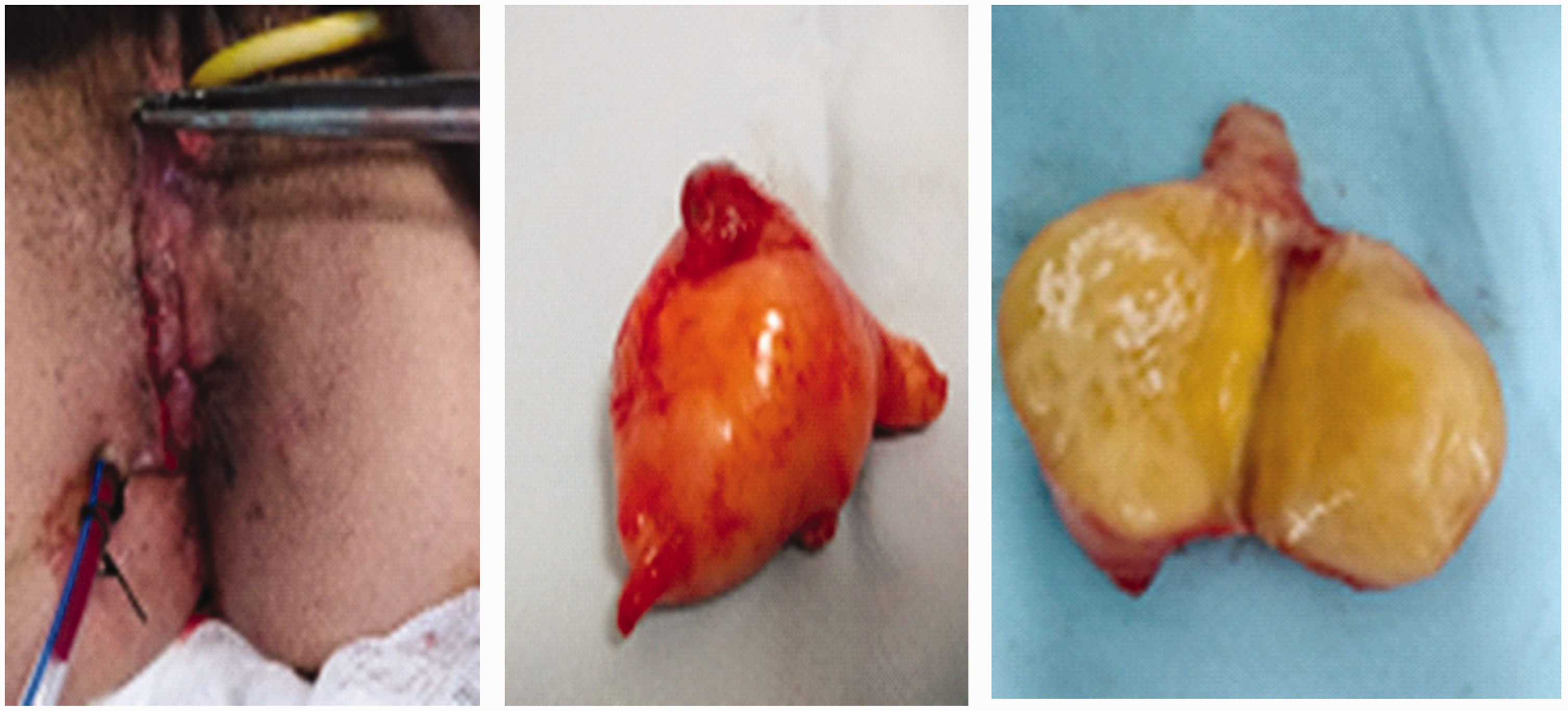
End of the procedure: para-rectal incision and surgical equipment.
A follow-up abdominal CT scan was performed 2 years postoperatively and showed a lesion resembling a schwannoma opposite to L1–L2 (Figure 5). This case was reported according to the CARE guidelines. 9 Consent for treatment and to publish this case were obtained from the patient.

Follow-up computed tomography scan showing an aspect resembling a radicular schwannoma (L1–L2).
Discussion
NF1 is the most commonly encountered form of tumor suppressor syndrome that predisposes patients to benign and malignant tumors. NF1 alone accounts for 96% of NF cases. 1 NF1 is a tumor suppressor gene that acts as a downregulator of the RAS/MAPK pathway through its GTPase activating protein-related domain, 10 which converts RAS-GTP to an inactive form, RAS-GDP 11 . RAS belongs to a signal transduction pathway involved in cell proliferation and differentiation. Therefore, any loss of neurofibromin function will lead to sustained levels of active RAS-GTP and, consequently, increased proliferation, uncontrolled growth, and loss of apoptosis. 8 As previously mentioned, the diagnosis is clinical and is based on the consensus of the National Institutes of Health. However, most often, the disease has a wide variety of manifestations that can make a proper diagnosis challenging.1,4 Even in families that carry the same mutation, a wide variability in clinical manifestations has been reported, making the establishment of definitive clinical criteria problematic. Other than the clinical criteria of the National Institutes of Health, a wide spectrum of clinical complications affecting several organs may accompany the disease. To date, genotype-phenotype correlations have only been found in patients with NF1 microdeletions or in those with a three-base-pair deletion in exon 17. In the former patients, the disease manifests with a higher number of dermal neurofibromas starting at a younger age, a lower IQ, and facial dysmorphism. 12 These patients generally have higher risk for malignant peripheral nerve sheath tumors. In contrast, patients with a three-base-pair deletion in exon 17 express a milder form of the disease, presenting with multiple café-au-lait macules and other features of NF1 but without cutaneous or plexiform neurofibromas. 13 Taken together, the combined intra-familial heterogeneity and the poor genotype-phenotype correlations may underscore, at least at present, the utility of genetic screening for mutations. However, some centers have started to perform genetic diagnoses and have identified many new mutations that may inform clinical decisions in the near future.14,15 This is very important not only for the diagnosis of new cases but also for preconception genetic counseling in families with NF1.
Our patient had four of the seven criteria: a first-degree family relative with a history of NF1, the presence of multiple café-au-lait spots, dorso-lumbar scoliosis, and a neurofibroma confirmed histologically a posteriori. The treatment consisted of surgical exeresis of the lesion, which resulted in an immediate favorable outcome. A histopathological examination revealed that the lesion was a benign neurofibroma.
Although benign in most cases, 8% to 15% of NF1 patients who develop neurofibromas progress to malignant peripheral nerve sheath tumors, and there are generally no predictors of this progression.16,17 This clinical dilemma increases the difficulty of determining how to best manage and monitor neurofibromas in NF1 patients to objectively intervene at an early stage and prevent the occurrence of an aggressive or metastasizing malignancy. 8 Considering this together with the possibility that the mass may be malignant or may progress to a malignant mass, especially in an NF1 patient, we opted for total surgical exeresis. In their 119 surgical cases, Artico et al. 18 also opted for and demonstrated that complete exeresis of neurofibromas and schwannomas is possible with an acceptable injury risk. This risk of injury, which may be increased in NF1 patients, should not preclude tumor removal because of the increased risk of malignant transformation in these patients. One other potential rational was that the patient’s mother had a history of surgical removal of a small intestine tumor associated with NF1. 18 A 2-year postoperative abdominal CT scan did not reveal any sign of recurrence but revealed the development of a new lesion resembling a schwannoma opposite to L1–L2. This finding illustrates the importance of regular and long-term follow-up of NF1 carriers.
Conclusion
NF1 or Von Recklinghaussen’s disease is an autosomal dominant disorder with a wide variety of manifestations. The disease is generally accompanied by multiple neurofibromas or schwannomas that may affect any nerve in the body. The risk of malignant transformation of these tumors is increased in patients with NF1, which justifies their resection. Although the risk of injuries associated with their removal is increased, here, we demonstrate a safe removal of a pararectal neurofibroma as evidenced by the absence of immediate postoperative neurological complications and 2-year follow-up recurrence. The risk of tumor occurrence and the unpredictable evolution of the disease necessitate regular and long-term follow-up of NF1 carriers. This is justified in this case report by the development of a completely new lesion.
Research Data
Research Data for A fortuitous discovery of a neurofibroma in a female patient with type 1 neurofibromatosis: a case report
Research Data for A fortuitous discovery of a neurofibroma in a female patient with type 1 neurofibromatosis: a case report by Fathia Harrabi, Jabeur Methnani, Ammar Houssem, Mizouni Abdelkader, Ben Latifa Mehdi, Saiid Mohamed Amine, Ben Mabrouk Mohamed and Ben Ali Ali in Journal of International Medical Research
Footnotes
Acknowledgement
The authors would like to acknowledge the patient for agreeing to publish this case.
Author contributions
FH, AH, and MA treated and clinically evaluated the patient; FH, MJ, and BLM drafted the manuscript. All authors contributed to the manuscript revision and approved the final version.
Data availability statement
Data pertaining to this case report are available from the corresponding author on reasonable request.
Declaration of conflicting interests
The authors declare that there is no conflict of interests.
Ethical approval
This case report was approved to be published by the local ethical committee of the Faculty of Medicine of Sousse (approval number: n°83/10-2022 provided on 5-10-2022). Written informed consent was obtained from this patient to publish this case.
Funding
This research was not funded by any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.