
Abstract
The bony cochlear nerve canal transmits the cochlear nerve as it passes from the fundus of the internal auditory canal to the cochlea. Stenosis of the cochlear nerve canal, defined as a diameter less than 1.0 mm in transverse diameter, is associated with inner ear anomalies and severe to profound congenital hearing loss. We describe an 11-month-old infant with nonsyndromic congenital sensorineural hearing loss with cochlear nerve canal stenosis. Next-generation sequencing revealed heterozygous mutations in MYH9 and MYH14, encoding for the inner ear proteins myosin heavy chain IIA and IIC. The patient’s hearing was rehabilitated with bilateral cochlear implantation.
Keywords
Introduction
The bony cochlear nerve canal (CNC) is located between the fundus of the internal auditory canal and the base of the cochlea. Cochlear nerve fibers, passing from the spiral ganglion to the cochlear nerve, must traverse the CNC. Cochlear nerve canal stenosis, which most commonly presents as congenital sensorineural hearing loss (SNHL), is associated with cochlear nerve anomalies and inner ear malformations. 1 These malformations typically arise during the first trimester of pregnancy secondary to disturbances in the formation of the membranous labyrinth. 2 Cochlear nerve canal stenosis has been estimated to be present in up to 18% of children with congenital SNHL and is associated with inadequate development of the cochlear nerve—either partial (hypoplasia) or complete (aplasia). 3
Various criteria for CNC stenosis have been proposed, including threshold diameters of 1.0, 1.2, or 1.4 mm. 2,4 The CNC is generally accepted as normal if the diameter exceeds 1.5 or 2.0 mm. 2 Smaller CNC diameters have been associated with poorer hearing performance both prior to and following cochlear implantation, 4,5 and objective electrophysiologic differences in patients with CNC stenosis undergoing cochlear implantation have also been described. 6 Individual genetic mutations associated with CNC stenosis have not previously been reported.
Myosin is a protein that plays an important role in a variety of cells and organ systems, and nonmuscle myosin heavy chain (NMMHC) is expressed in the inner ear. Mutations in MYH9 and MYH14, encoding for NMMHC-IIA and IIC, respectively, are associated with autosomal dominant nonsyndromic hearing loss. 7 In this report, we present an interesting case of congenital SNHL and CNC stenosis with the proband demonstrating single mutations in both the MYH9 and the MYH14 gene.
Case Report
Institutional review board approval and parental/patient consent were obtained for this study. A 6-month-old female patient presented to a tertiary otolaryngology center with bilateral hearing impairment since birth; the patient failed newborn hearing screening examinations. There was no family history of hearing loss, and the parents were of normal hearing. An audiogram confirmed bilateral profound SNHL. Physical examination revealed normal external auditory canals and tympanic membranes bilaterally.
Further testing included auditory steady-state response (ASSR), computed tomography (CT), and magnetic resonance imaging (MRI). Genetic analysis was then performed on the proband and parents. This consisted of a NimbleGen chip for target enrichment followed by next-generation sequencing (Roche Sequencing) to detect for 127 genes associated with hearing loss, including the 2 most common mutations seen in nonsyndromic autosomal recessive hearing loss, GJB2 and GJB6. 8 Interpretation of sequencing results was followed by bioinformatic analysis using international mutation and polymorphism databases, including Sorting Intolerant From Tolerant, 9 which allowed for assessment of the mutations and clinical correlation.
Audiological testing results were as follows: ASSR revealed thresholds of 100 dB in the right ear at 500, 1000, 2000, and 4000 Hz; the left ear tested at 90, 100, 100, and 105 dB, respectively. Distortion product otoacoustic emissions were absent bilaterally. A 40 Hz auditory event-related potentials revealed a hearing threshold of 90 dB in the right ear and 100 dB nHL in the left ear. Acoustic imittance examination was normal, with type A tympanograms bilaterally.
High-resolution CT of the temporal bones was performed on coronal and axial planes. This revealed dysplasia of the cochleae, vestibules, and semicircular canals bilaterally, with bilateral CNC stenosis and mildly enlarged vestibular aqueducts, most closely resembling a Mondini malformation or incomplete partition type II (Figure 1). 10 Magnetic resonance imaging was performed, revealing congenital absence of the cochlear nerves bilaterally (Figure 2).
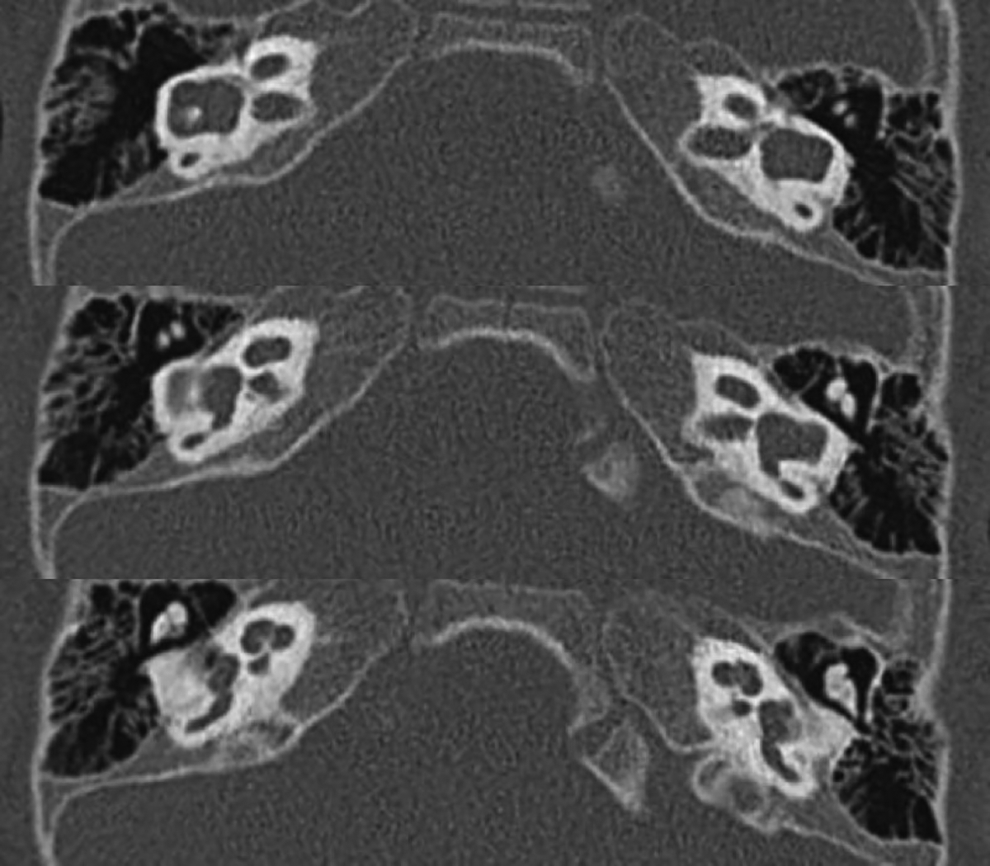
Computed tomography reveals bilateral cochlear nerve canal stenosis in addition to cochleae with cystic apices, dilated vestibules, and mildly enlarged vestibular aqueducts. The inner ear malformations most closely resemble a Mondini malformation.
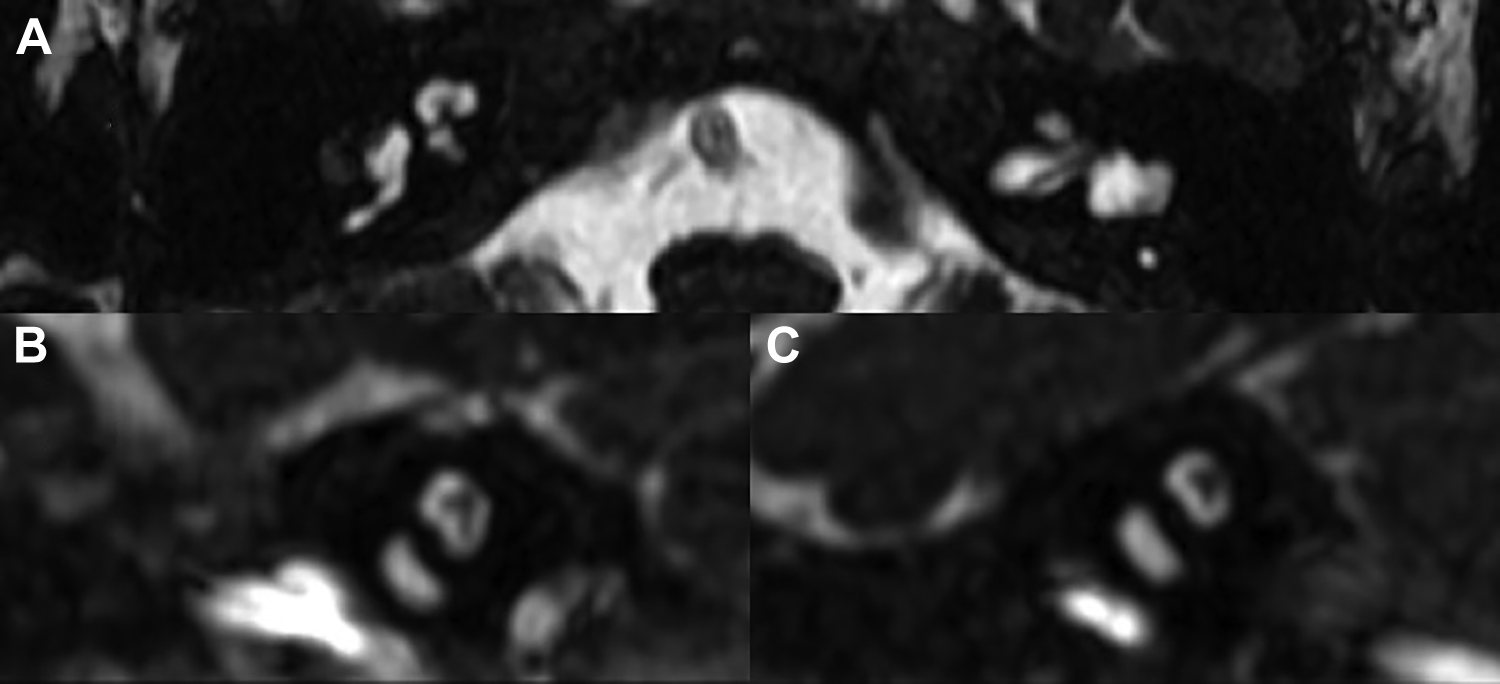
Magnetic resonance imaging, heavily T2-weighted sequence, reveals bilateral cochlear nerve aplasia. A, Axial view. B, Sagittal view, left side. C, Sagittal view, right side.
Positive results were noted in 2 genes, the first of which was MYH9 (NM_002473; c.41A>C; p.Lys14Thr|p.K14 T; CDS1; heterozygous; missense). There has been no report to date on the pathogenicity of this locus; the probability of occurrence of this locus in the patient’s native population, the Chinese population, is deemed to be low. Sorting Intolerant From Tolerant predicted that protein function was most likely to be affected. MYH9 mutations result in autosomal dominant, nonsyndromic hearing loss type 17. The second gene demonstrating a mutation was MYH14 (NM_001077186; c.3346G>A; p.Glu1116Lys|p.E1116 K; CDS25; heterozygous; missense). There has been no report to date on the pathogenicity of this locus; the probability of occurrence of this locus in the Chinese population is deemed to be low. Sorting Intolerant From Tolerant was used to predict pathogenicity, and this suggested that protein function was most likely affected. MYH14 mutations result in autosomal dominant, nonsyndromic heating loss type 4A.
Genetic testing was performed on the proband’s parents. Interestingly, analysis revealed that the proband inherited MYH9 (c.41A>C, p.Lys14Thr) from her normal hearing father, and MYH14 (c.3346G>A, p.Glu1116Lys) from her normal hearing mother.
Given these findings, the patient was fitted with bilateral hearing aids, but this resulted in no significant rehabilitation of her bilateral profound loss. At age 11 months, the patient underwent bilateral cochlear implantation (MED-EL SONATA FLEX28) via a standard cochleostomy approach, and full insertion was obtained intraoperatively. Intraoperative evoked auditory nerve telemetry, which has been associated with higher stimulation thresholds required in patients with CNC stenosis, 6 produced responses in several electrodes in the apex of the cochleae bilaterally, confirming intracochlear placement of the electrode.
Two months following activation, the patient demonstrated Categories of Auditory Performance (CAP) 3/9, speech intelligibility rating (SIR) 1/5, and Infant-Toddler Meaningful Auditory Integration Scale (IT-MAIS) 24/40. Three months following activation, performance was graded at CAP 4/9, SIR 1/5, and IT-MAIS 25/40, representing significant rehabilitation of hearing.
Discussion
Cochlear nerve canal stenosis, first described by Fatterpekar in 2000, is defined as a bony CNC less than 1.0 mm in transverse diameter on CT imaging; a normal CNC is greater than 1.5 mm in diameter. 2 Cochlear nerve canal stenosis is associated with inner ear malformations and cochlear nerve hypoplasia or aplasia. If CNC stenosis is diagnosed, MRI examination may elucidate the presence or absence of a cochlear nerve within the internal auditory canal. Although cochlear nerve aplasia has traditionally been described as a contraindication to cochlear implantation, 11 interestingly, some patients with cochlear nerve aplasia experience hearing benefit after implantation. This may be explained by the presence of cochlear nerve fibers traveling along the other nerves, such as the vestibular nerves, in the internal auditory canal. 12
Congenital hearing loss may arise secondary to perinatal infections or genetic mutations. The most common nonsyndromic genetic cause of congenital hearing loss is a mutation in GJB2 or GJB6, which encode for gap junction (connexin) proteins in the inner ear. These mutations are typically associated with morphologically normal inner ear structures and internal auditory canals. The most common syndromic congenital hearing loss presentations are Pendred syndrome, caused by mutations in SLC26A4, and Usher syndrome, a heterogeneous disease caused by mutations in up to 16 different loci on various chromosomes. 13,14 Patients with Pendred syndrome can present with an enlarged vestibular aqueduct and experience progressive hearing loss. Similar, patients with Usher syndrome usually experience progressive hearing loss, although there is wide phenotypic variation. Aside from GJB2 and SLC26A4, a prominent genetic hearing loss etiology is believed to be the wider category of mitochondrial mutations, which can cause both syndromic and nonsyndromic patterns of hearing loss. 15
In contrast, CNC stenosis is relatively rare, and, to our knowledge, has not previously been reported to be associated with specific genetic mutations. In this report, an infant with bilateral profound hearing loss was diagnosed with mutations in MYH9 and MYH14, each of which has separately been associated with autosomal dominant, nonsyndromic hearing loss. 16 These 2 genes encode for NMMHC IIA and IIC, respectively, and are expressed in the inner ear. MYH9 mutations are associated with MYH9-related disease, including May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome, lying along a spectrum including thrombocytopenia, SNHL, nephropathy, cataracts, and liver abnormalities. 7,17 Our patient did not express any syndromic phenotype. In the inner ear, NMMHC-IIA is expressed in the organ of Corti, the subcentral region of the spiral ligament, and Reissner’s membrane. 18 Similarly, MYH14 is located within the autosomal dominant hearing impairment critical region, 16 and its product NMMHC-IIC has been reported to play a beneficial role in cochlear protection following acoustic overstimulation in a mouse model. 19 Two single-nucleotide polymorphisms in MYH14 (rs667907 and rs588035) have been associated with hearing loss in a prior report. 20 Neither gene has been associated with CNC stenosis.
Interestingly, in this case, the proband inherited the MYH9 mutation from her mother and the MYH14 mutation from her father. As her parents were of normal hearing, it may be inferred that the compounded effect of these 2 mutations likely contributed to her unique phenotype of congenital hearing loss, inner ear malformation, and CNC stenosis.
Since initiation of next-generation sequencing at our institution for congenital hearing loss, 30 additional patients have been tested, and no other MYH9 or MYH14 mutations have been detected. These 2 genes are unlikely to be the only genes associated with CNC stenosis, and further research will concentrate on investigating other potential genetic etiologies.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.