
Abstract
MYH9 is a gene that encodes for a subunit of the myosin heavy chain IIA protein. Mutations in MYH9 are associated with hematologic abnormalities, renal dysfunction, and hearing loss. Bony cochlear nerve canal stenosis (CNCS), which is diagnosed on computed tomography (CT) imaging, has been associated with congenital deafness, cochlear nerve aplasia/hypoplasia, and inner ear malformations. We report two cases of CNCS presenting with profound congenital hearing loss whom we diagnosed with mutations in MYH9 and discuss the genotype-phenotype association and implications for management.
Introduction
Stenosis of the bony cochlear nerve canal (CNC), or CNCS, has been associated with congenital hearing loss. It is defined as a transverse diameter of the bony cochlear nerve canal (CNC), as measured on computed tomography (CT), of less than 1.0 mm, and has been associated with hypoplasia or aplasia of the cochlear nerve and inner ear malformations. The exact mechanism for CNCS is unknown, but proposed mechanisms include acquired stenosis versus failure of canalization.1,2
CNCS is typically diagnosed in the setting of a congenital profound hearing loss, which may be unilateral or bilateral. 3 Syndromic associations are rare, although the incidence of accessory auricles in patients with CNCS seems to exceed that encountered purely by chance. Cochlear implantation (CI) can lead to significant rehabilitation of hearing, but patients with cochlear nerve aplasia on magnetic resonance imaging (MRI) should ideally be identified preoperatively. 4
The MYH9 gene lies on chromosome 22q11-13 and encodes non-muscle myosin heavy chain IIA (NMMHC-IIA). 5 Class IIA non-muscle myosin is a ubiquitous protein that is important for key cellular processes, including adhesion, cell migration, cytoplasmic division and polarization, maintenance of cell shape, and signal transduction. 6 Mutations in MYH9 are associated with MYH9-related disease, whose manifestations include hematologic, renal, ophthalmologic, and hepatic abnormalities, in addition to hearing loss.
We have previously reported an association between CNCS and mutations in MYH14 and MYH9 genes in a single patient. 7 In this follow-up investigation, we describe, more specifically, an association between CNCS and heterozygous MYH9 mutations in two patients, and describe implications for management.
Case Reports
Institutional review board approval was obtained for this study (IRB no. 2021-P2-044-01) and informed consent was obtained. Two patients with CNCS were identified. Imaging studies performed included CT of the temporal bones and MRI of the brain and internal auditory canals. Genetic analysis was performed on both patients in the form of a NimbleGen chip for target enrichment followed by next generation sequencing (Roche Sequencing, Pleasanton, CA). Testing was performed for 127 genes associated with hearing loss, including GJB2 and GJB6, the two most common mutations seen in nonsyndromic autosomal recessive hearing loss. Bioinformatics analysis was then performed using international mutation and polymorphism databases, including Sorting Intolerant from Tolerant (SIFT).
Audiological testing included auditory steady-state response (ASSR), auditory brainstem response (ABR), 40-Hz auditory event-related potentials (AERP), and distortion product otoacoustic emissions (DPOAE). Radiological studies performed included CT and magnetic resonance imaging (MRI).
Patient 1
This case was summarized in a previous report. 7 A 6-month-old female presented with bilateral profound hearing loss. Pregnancy and early infancy were unremarkable with the exception of jaundice, which resolved spontaneously. There was no history of otitis media. Family history was negative for hearing loss. The infant failed the newborn hearing screen, and parents noted that she was poorly responsive to sounds during the first year of life. Bilateral hearing amplification was fitted at 7 months of age.
Physical examination revealed normal external ears and tympanic membranes bilaterally. Audiological testing results were as follows. Bilateral type A tympanograms were present. ASSR showed thresholds of 100 dB nHL in the right ear at 500, 1000, 2000, and 4000 Hz; the thresholds in the left ear were 90, 100, 100, and 105 dB nHL at each frequency. 40 Hz AERP showed hearing thresholds of 90 dB nHL in the right ear and 100 dB nHL in the left ear. ABR showed undetectable waves at 97 dB nHL in both ears. DPOAE was absent across all frequencies bilaterally.
CT revealed bilateral CNCS (complete stenosis, diameter 0 mm) and mildly enlarged vestibular aqueducts, resembling Mondini malformation/incomplete partition type II. Bilateral cochlear nerve aplasia was noted on MRI.
Genetic analysis was performed as described above. Two mutations of note were discovered: the first, in MYH9 (NM_002473; c.41A>C; p.Lys14Thr|p.K14T; CDS1; heterozygous; missense), and the second, in MYH14 (NM_001077186; C.3346G>A; p.Glu1116Lys|p. E1116K; CDS25; heterozygous; missense).
Following presentation to the tertiary medical center, a trial of hearing amplification was first implemented, with no measurable benefit. An informed consent discussion regarding cochlear implantation was undertaken with the patient’s parents. The risk of nonstimulation with a cochlear implant were discussed, and the parents agreed to proceed with cochlear implantation. At 11 months of age, the patient underwent bilateral cochlear implantation (MED-El Sonata FLEX28) through a standard cochleostomy approach; full insertion was obtained. Intraoperative electrocochleography was performed, and responses were present in several electrodes bilaterally toward the apex of the cochlea.
Two months following activation of the implant, the patient exhibited Categories of Auditory Performance (CAP) 3/9, Speech Intelligibility Rating (SIR) 1/5, and Infant-Toddler Meaningful Auditory Integration Scale (IT-MAIS) 24/40. Three months after use, the child demonstrated continued progress, with CAP 4/9, SIR 1/5, and IT-MAIS 25/40. No longer-term follow-up was available.
Patient 2
A 15-year-old male presented to the tertiary medical center with subjectively poor hearing in his right ear since childhood; the left ear was asymptomatic, and hearing was subjectively normal on the left. Pregnancy, infancy, and early childhood were unremarkable. There was no history of otitis media.
Physical examination revealed normal external ears and tympanic membranes bilaterally. Audiological testing results were as follows. Pure-tone audiometry revealed normal hearing thresholds on the left and profound hearing loss on the right. Bilateral type A tympanograms were present. ASSR revealed thresholds of 80, 100, and 100 dB nHL at 500, 1000, and 2000 Hz for the right ear, respectively, with no response at 4000 Hz; the thresholds for the left ear were 20, 30, 20, and 20 dB nHL at the same frequencies, respectively. 40 Hz AERP showed hearing thresholds of 80 dB nHL in the right ear and 20 dB nHL in the left ear. DPOAE were absent on the right and normal on the left.
CT revealed CNCS on the right (diameter 0.6 mm) and a normal bony CNC on the left. MRI confirmed these results, and the cochlear nerve was present bilaterally. Genetic analysis was performed as described above. One mutation of note was discovered: in MYH9 (NM_002473; c.2449G>T; p.Ala817Ser; heterozygous; missense).
The patient and family were offered a unilateral cochlear implant, but they chose not to proceed. The patient did not return for further follow-up.
Discussion
Cochlear nerve canal stenosis (CNCS), which may be present unilaterally or bilaterally, is typically associated with congenital hearing loss. The hearing loss encountered is usually profound. Associated developmental abnormalities include hypoplasia or aplasia of the cochlear nerve as well as inner ear malformations.
8
Various thresholds for normal cochlear nerve canal diameter have been proposed; the most common thresholds cited are either 1.5 mm or 1.0 mm, with some authors suggesting other values (including 1.7 to 1.8 mm).3,7,9,10 The degree of stenosis is correlated with the likelihood of cochlear nerve aplasia or hypoplasia, with at least two studies suggesting that a CNC diameter of under 1.5 mm is highly associated with aplasia/hypoplasia (Figure 1).11,12
A, computed tomography (CT), axial view. Right cochlear nerve canal stenosis is seen (arrow). B, left bony cochlear nerve canal is normal (arrow). C, magnetic resonance imaging (MRI), axial view. Normal inner ear and internal auditory canal morphology noted. D, E, MRI, sagittal view. Nerve elements noted on cross-sectional view (arrows).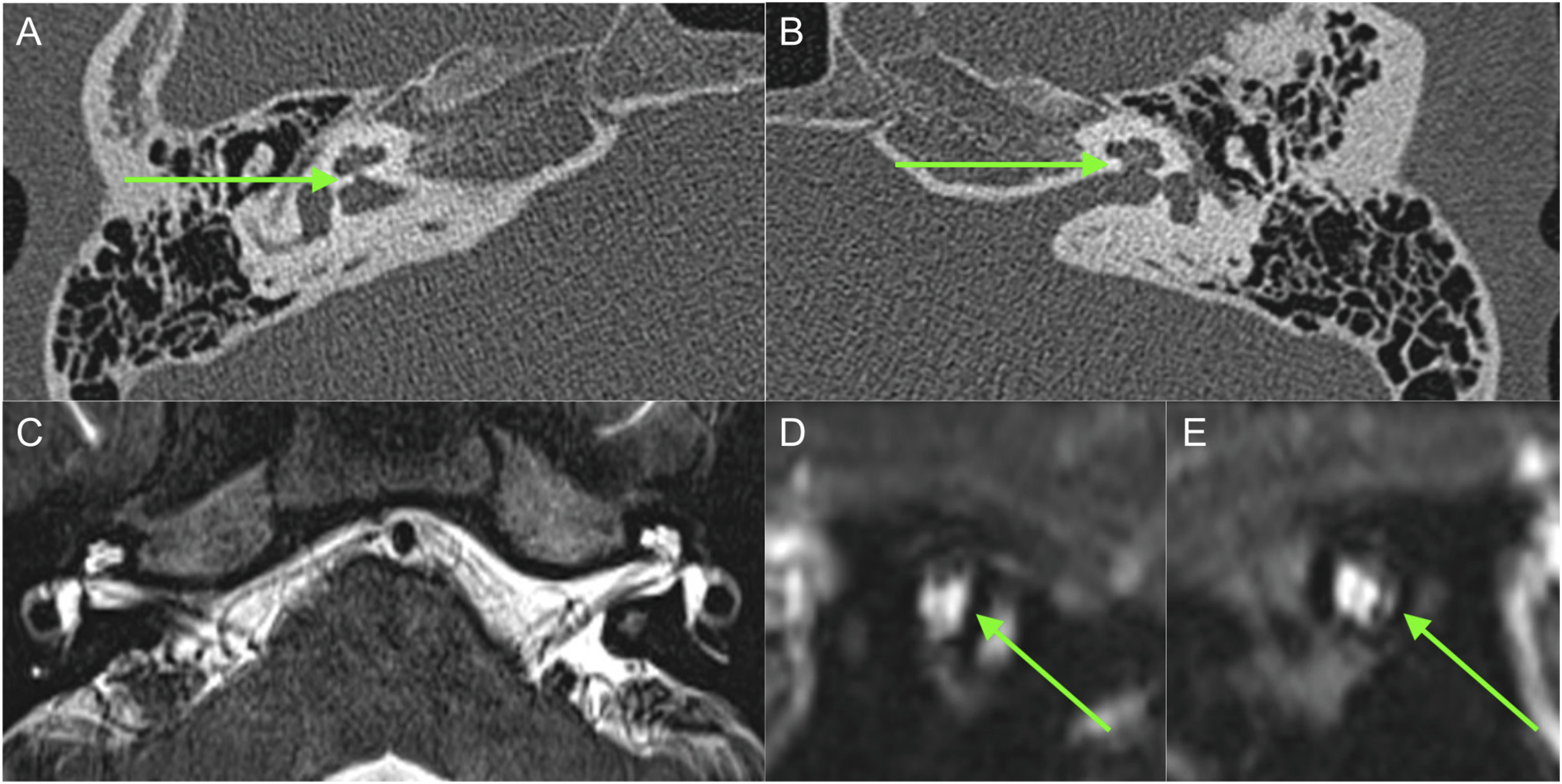
As a protein superfamily, myosins use the energy generated during ATP hydrolysis to drive conformational changes, thus acting as molecular motors. The motor domain of class II myosins binds to actin filaments to form fine filaments and generate force and tension. Class II myosins can be broadly classified as muscle (skeletal and cardiac), smooth muscle, and non-muscle. Non-muscle class II myosins are divided three variants based on their heavy chain. The MYH9 gene encodes one of them, NMHC IIA (or NM IIA). This is a gene with 40 coding exons, containing an untranslated exon 1a at the 5' end. 13 Exons 1 to 18 translate to the globular head, exon 19 translates to the neck, and exons 20 to 39 are expressed as discoids. Finally, the non-helical structural tail at the C-terminus is translated from exon 40. Heavy chain proteins include an N-terminal structural domain, which in turn includes a globular head structural domain (or motor structural domain) that interacts with actin and hydrolyzes ATP, and a tail structural domain that acts as a lever arm for the motor structural domain and as a binding site for the light chain. The tail structural domain contains a long α-helix discoidal tubular domain and a short carboxy-terminated helical portion. 14
The MYH9 gene is widely expressed in human and mouse cochlear tissues, especially in the hair cell stereocilia, and it has been hypothesized that mutations in MYH9 gene disrupt the structural integrity of hair cell stereocilia, leading to deafness in mouse models. 15 A mouse model expressing the p.R702C heterozygous mutation by knock-in method exhibited SNHL. 16 Head structural domain mutations are more likely to cause hearing loss than tail structural domain mutations, and patients with head structural 17 domain mutations almost always experience hearing loss before the age of 40 years. 18 Hearing loss caused by MYH9 mutations is more common in children or adolescents. 19 This is consistent with the results of this study, where both subjects with discovered mutations in these genes had hearing loss at a young age.
Besides otologic manifestations, MYH9 has been implicated in other disorders, including, most notably, the condition termed MYH9-related disease (MYH9-RD). MYH9-RD is associated with hematologic abnormalities in addition to renal dysfunction, presenile cataracts, and liver function abnormalities. MYH9-RD encompasses a variety of phenotypes, including Epstein syndrome, Fechtner syndrome, May-Hegglin anomaly, and Sebastian syndrome. 6
Other specific mutations have been described in the MYH9 gene. Wu et al. found that a p.E1256K mutation in the MYH9 gene caused nonsyndromic SNHL. 20 Dantas et al. reported a p.R705H mutation as the causative gene of DFNA17 hearing loss. 21 The phenotype also varies. Verver et al. reported a family with a p.R705H mutation in MYH9 in which all four subjects had symmetric, high-frequency hearing impairment followed by rapid progression to very severe full frequency hearing impairment with age. 17 Wasano et al. reported a p.D1424N mutation in a family with five subjects with MYH9 gene mutation in which the primary subject and the subject’s father demonstrated progressive hearing loss, leading to a bilateral profound loss. 22 The primary subject was diagnosed with hearing loss by the age of 29 years (bilateral moderate-to-severe high-frequency hearing loss and mild low and mid-frequency hearing loss). The father of the subject was diagnosed with hearing loss at age 40, characterized by high-frequency hearing impairment, and progressed to profound deafness at age 54.
Although the above studies have shown an association between MYH9 and SNHL, no study has reported an association between MYH9 and CNCS aside from our previously published account. 7 The association remains clear, but it remains to be demonstrated whether the phenotype of CNCS in the patients in this study is a casual association, caused by genetic abnormalities in the MYH9 gene.
At present, the principles of treatment of patients with profound hearing loss associated with mutations in MYH9 are the same as those for patients with nonsyndromic congenital profound loss, including amplification and CI, when appropriate. CI has been demonstrated to be effective in patients with MYH9-related disease, although the presence or absence of CNCS was not specifically studied.23,24 Patient 1 in this study underwent CI and demonstrated good hearing recovery 3 months after surgery; patient 2 refused further treatment, such as CI, for the unilateral profound right hearing loss, and did not present for further follow-up.
Conclusion
We report 2 pediatric cases of hearing loss presenting with bony cochlear nerve canal stenosis (CNCS) in the presence of MYH9 gene mutations. Imaging and audiological examinations confirmed the diagnosis of CNCS, and both genetic tests revealed heterozygous mutations in MYH9. However, further genetic analysis in other CNCS patients is needed to increase our understanding of the genetic factors in the pathogenesis of CNCS. Due to the genetic complexity of CNCS, there is an existing need to perform genetic analysis in CNCS patients, and such testing should be considered when evaluating a patient with CNCS. Furthermore, CT imaging for CNCS should be considered in patients with DFNA17 or with MYH9-related disease presenting with hearing loss.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.