
Abstract
Granular cell tumor (GCT) is a rare benign soft tissue neoplasm that develops primarily in the skin and subcutaneous tissue of the head and neck, frequently in the tongue, but has not been observed in the auditory meatus. These tumors generally present as slow-growing nodules with few unique clinical or radiological features, and so must be confirmed by pathological examination. Here, we present the case of a 62-year-old female with right hearing loss and benign GCT in the right auditory meatus initially misdiagnosed as cholesteatoma.
Introduction
Granular cell tumor (GCT) is classified as a benign soft tissue neoplasm according to the World Health Organization. 1 It can arise anywhere in the body, including the torso and limbs, but is most commonly found in the skin and subcutaneous tissues of the head and neck, particularly in the tongue. In fact, tongue lesions account for about one-fourth of all GCT cases. Sporadic cases have also been reported in the respiratory tract, breast, gastrointestinal tract, and vulva, 2 -4 but no cases of GCT have been documented in the auditory meatus.
Granular cell tumor can develop at any age but is most frequent in individuals 40 to 70 years with modest female preponderance. 1 It usually appears as painless papules or nodules lacking specific clinical symptoms. It also resembles many other neoplasms such as lipoma, fibroma, neuroma, and schwannoma. It is thus difficult to confirm GCT based on clinical manifestations or imaging results, although it is comparatively easy to diagnose by histopathology. Further, if occurring in the auditory meatus, it must be distinguished from cholesteatoma.
In this article, we present the first reported case of benign GCT in the auditory meatus, which was first misdiagnosed as cholesteatoma. Written informed consent was obtained from the patient for publication of this case report.
Clinical Summary
A 62-year-old female visited our hospital with a painless mass in the right auditory meatus. She reported a right ear itch of no discernible cause for the past 10 years, and frequent scratching led to bleeding and discharge of yellowish-white pus. Hearing in the right ear also gradually declined. About 4 years ago, she found a mass in the right auditory canal that increased gradually in diameter and also bled and discharged pus. In the past year prior to the current presentation, the patient also developed a tic in her right cheek that gradually increased in severity. Ear physical examination revealed no deformity in bilateral outer ears and no dragging pain in bilateral auricles. However, a faint, smooth, red lump about 0.5 cm in diameter and 1 cm in height blocked the right auditory canal, while the left auditory canal was clear. Further, left ear hearing was normal. A computed tomography scan of the right ear revealed chronic otitis media with suspected cholesteatoma (Figure 1).
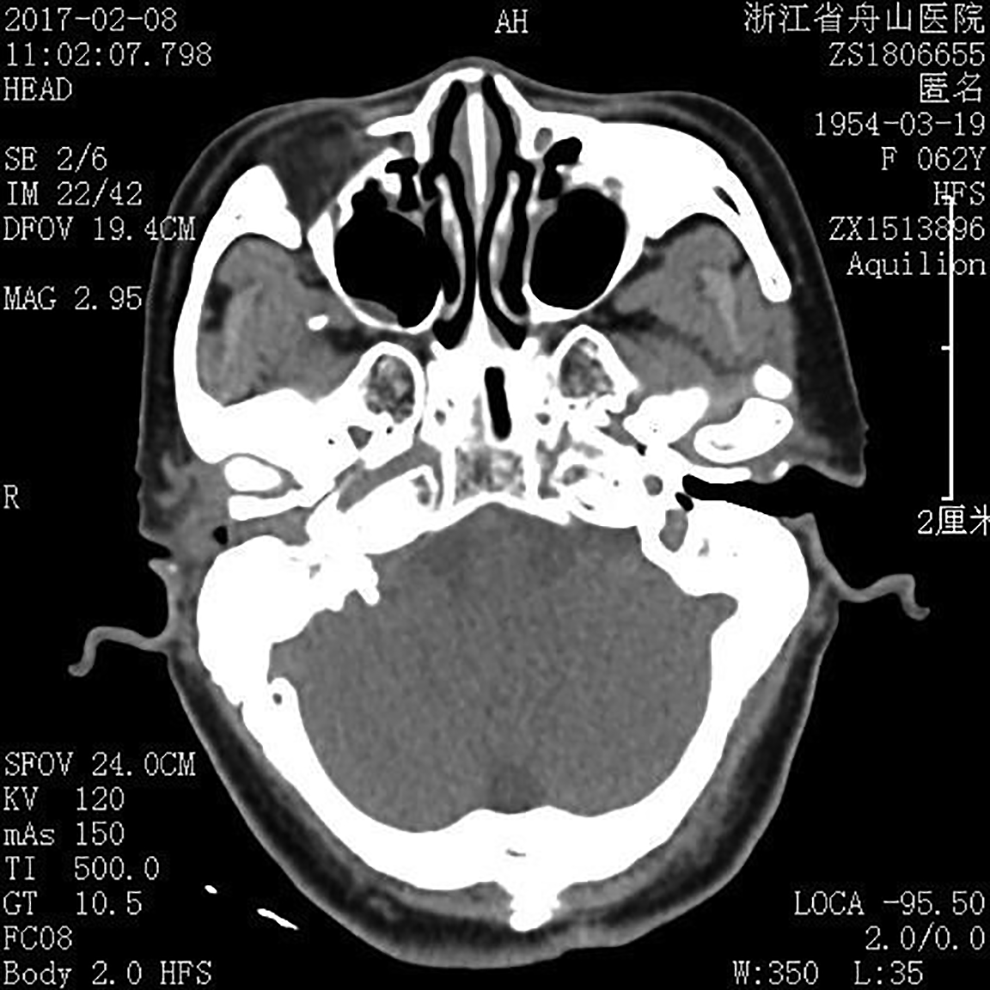
Computed tomography (CT) scan of the right ear showing chronic otitis media and suspected cholesteatoma.
Pathological Findings
The entire lesion was excised and the specimen fixed in 10% buffered formalin for histopathological analysis. On gross examination, the mass was brown and measured 10 × 10 × 5 mm. Under light microscopy, round or polygonal cells were arranged into nests, sheets, or wide stripes. The tumor cell mass was covered with squamous epithelium and separated by fibrous connective tissue. Individual tumor cells were characterized by small hyperchromatic central nuclei and voluminous eosinophilic, granular cytoplasm with no signs of mitosis (Figure 2).
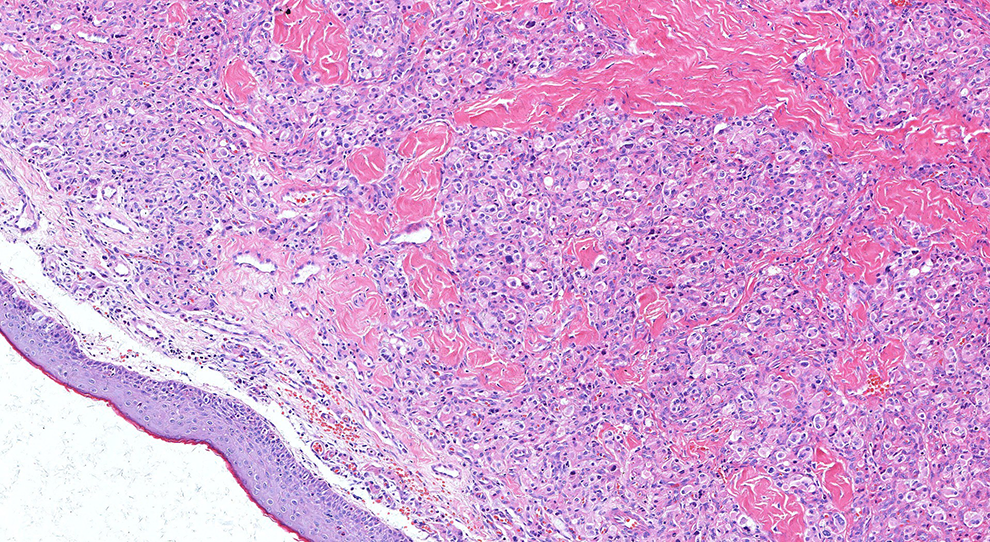
Hematoxylin and eosin (HE)–stained biopsy sample obtained from the patient (magnification, 200×).
To better classify the tumor, an immunohistochemical staining panel including S-100, CK, neuron-specific enolase (NSE), calretinin, inhibin, Melan-A, HMB45, and Ki-67 was performed using the Streptavidin–Biotin complex technique. The tumor cells were diffusely but strongly reactive for S-100 (Figure 3) and NSE (Figure 4), but negative for calretinin, inhibin, Melan-A, and HMB45. The Ki-67 index was <5% (Figure 5). All slides were reviewed and reevaluated independently by 3 pathologists.
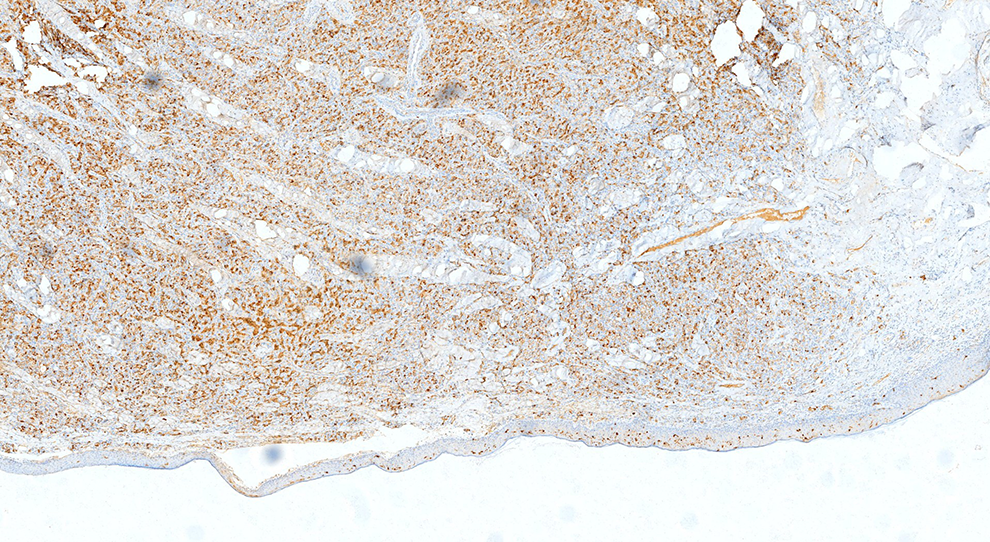
Immunohistochemical staining for S-100 (magnification, 40×).
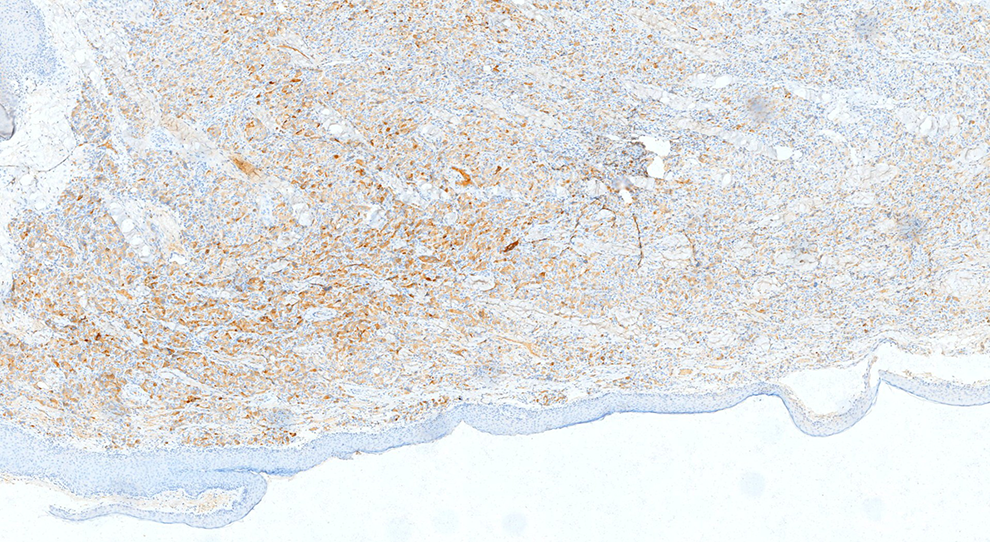
Immunohistochemical staining for neuron-specific enolase (NSE; magnification, 200×).
Immunohistochemical staining for Ki-67 (magnification, 40×).
According to these histological features and immunophenotypes, the mass was confirmed as GCT. The patient was discharged when all vital signs were stable. No mass recurrence has been detected during the 6-month follow-up period.
Discussion
Granular cell tumors are comparatively uncommon and benign neoplasms first described by Abrikossoff in 1926 as “granular cell myoblastoma.” 5 About 30% of all cases occur in the tongue, and a large proportion of the remaining cases are found in other soft tissues of the head and neck, while GCTs are rare in breast, proximal limbs, and internal organs. 6 To the best of our knowledge, no case of GCT in the auditory meatus has been reported. The most common tumor of the auditory meatus is cholesteatoma, which is usually associated with inflammation. Cholesteatoma is a cystic expanding lesion lined by stratified squamous epithelium containing a large amount of desquamated keratin. 7 When cholesterol crystals precipitate out of the insoluble keratin, it can aggravate local irritation and initiate inflammation. Considering that the patient had a long history of otitis media, the radiologists and surgeons initially misdiagnosed the mass as cholesteatoma.
Neither GCTs nor cholesteatoma have unique signs and symptoms for differential diagnosis. Both grow slowly and present as mobile painless masses, although they are easily distinguished by histopathology. Granular cell tumors vary from <1 to 12 cm in diameter, but in most cases, the nodule is smaller than 3 cm3 as in the current case (∼1 cm in diameter). In histopathological sections, GCTs show an abundance of relatively large polygonal or elongated tumor cells growing in sheet- and nest-like patterns surrounded by collagen fibers. Individual cells usually exhibit voluminous cytoplasm containing thick acidophilic granules and a single small round nucleus. The overlying squamous epithelium may be hyperplastic with keratin pearls visible in some cases. 1
The etiology of GCT is still unclear. Abrikossoff speculated that they are derived from striated muscle, while Ordóñez proposed that GCTs are of neurogenic origin with characteristics of Schwann cell differentiation, as the tumor cells are usually immunoreactive for S-100, NSE, and CD68. 8
Although GCTs lack clinical specificity, they have special morphological characteristics that combined with immunohistochemical expression patterns allow for definitive identification. However, the histopathological structure of malignant GCT, which accounts for about 2% of all GCT cases, is similar to benign GCT and there is no established histopathological standard for differentiation. Fanburg-Smith et al 9 proposed that certain histological features such as necrosis, excessive spindle cell number, vesicular nuclei with prominent nucleoli, increased mitotic activity (>2 mitoses/10 high-power fields), high nucleocytoplasmic ratio, and pleomorphisms were associated with increased risk of metastasis and that 3 or more of these signs are strongly indicative of malignancy. In addition, tumor diameter, growth rate, and metastasis can distinguish malignant from benign GCT.
The current recommended treatment for GCT is complete surgical excision, and prognosis is considered good following removal. Local excision should be wide as GCTs are not encapsulated and may infiltrate the surrounding tissue.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.