
Abstract
Non-neural granular cell tumor was first described in 1991 as an unusual primitive, polypoid variant of the conventional granular cell tumor. To date, this neoplasm remains a rare entity and the cell of origin is uncertain. While the histological features are similar to the conventional granular cell tumor, it represents a distinct entity that is negative for S100 and lacks true nerve sheath differentiation. Here, we describe a case of a 4-year-old male who presented with a painless, soft nodule on his right chest wall that was slowly increasing in size. The mass was excised and sent for pathologic analysis. Microscopic examination reveals spindle and epithelioid cells with vesicular nuclei and prominent granular eosinophilic cytoplasm. Immunohistochemical analysis shows negative staining for S100 and AE1/AE3/PCK26 but is positive for CD68. A diagnosis of a non-neural granular cell tumor was made. We report a rare and diagnostically challenging case in a pediatric patient.
Introduction
Primitive non-neural granular cell tumor (PNGCT) is a rare tumor that was first described by LeBoit et al. 1 in 1991. He described four patients with tumors that showed a polypoid, expansile configuration. The histologic features were distinct from the conventional granular cell tumor and included numerous mitotic figures and cytologic atypia. He defined these entities as a ”primitive polypoid granular-cell tumor.” Since his original discovery of this neoplasm, it has been reported only rarely in the literature. PNGCT most frequently occurs in young adults and adolescents, with few cases in patients younger than 5 years of age. The tumor typically presents as a smooth, soft nodule arising in the dermis occurring most frequently on the trunk, extremities, or head and neck regions.2,3
Although PNGCT shares similar histologic features with the conventional granular cell tumor, as LeBoit demonstrated, it can be distinguished histologically by increased mitotic activity, nuclear pleomorphism, vesicular nuclei, and prominent nucleoli. In addition, PNGCT is S100 negative due to the absence of nerve sheath differentiation. 2
Case report
A 4-year-old male presented with a painless, soft polypoid small nodule on his right chest wall.
The nodule had a red to tan surface and, for the last 3 months, had grown rapidly from a pinpoint lesion to a 0.9 cm × 0.8 cm × 0.7 cm nodule. Ultrasound imaging showed a heterogeneous hypoechoic lesion with no definitive internal vascularity, most suggestive of a hematoma. Due to the increasing size of the lesion, the mass was excised.
Histological examination reveals a well-circumscribed lesion consisting of spindle and epithelioid cells that show prominent granular eosinophilic cytoplasm and vesicular nuclei (Figures 1–3). Cytologic atypia is not identified. The tumor extends to the subcutaneous adipose tissue, but the margins of resection are negative. Immunohistochemical staining shows tumor cells staining positive for CD68 and negative for S100 and AE1/AE3/PCK26 (Figures 4 and 5). The SOX10 and NSE are negative (Figures 6 and 7). Postoperative follow-up of our patient at 44 months showed no evidence of lymphadenopathy or reoccurrence of the tumor.
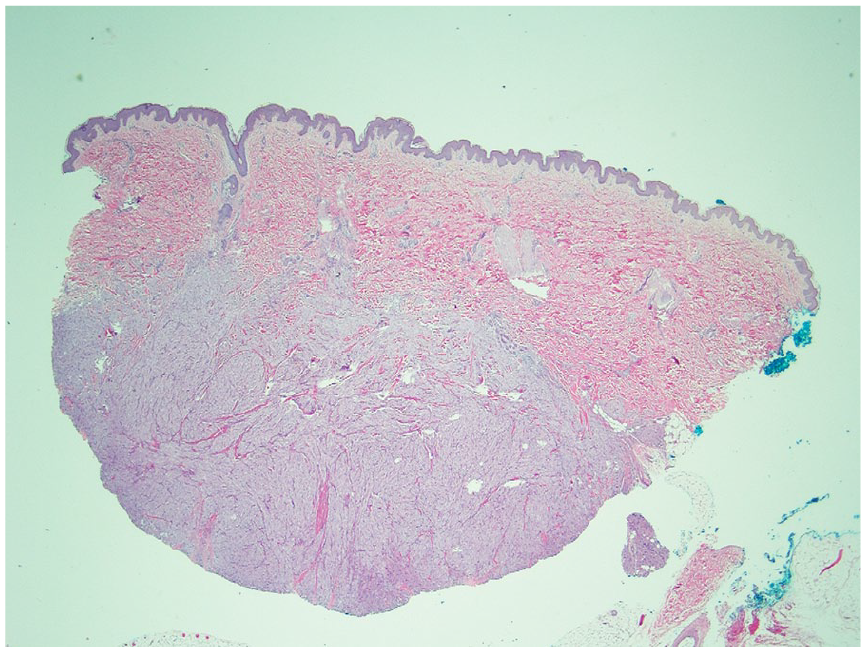
Dermal nodule with well-circumscribed but unencapsulated margin.

Epithelioid cells with prominent granular eosinophilic cytoplasm (10×).

Spindle and epithelioid cells with prominent granular eosinophilic cytoplasm (40×).

CD68 is positive in granular cells.

S100 is negative in granular cells.
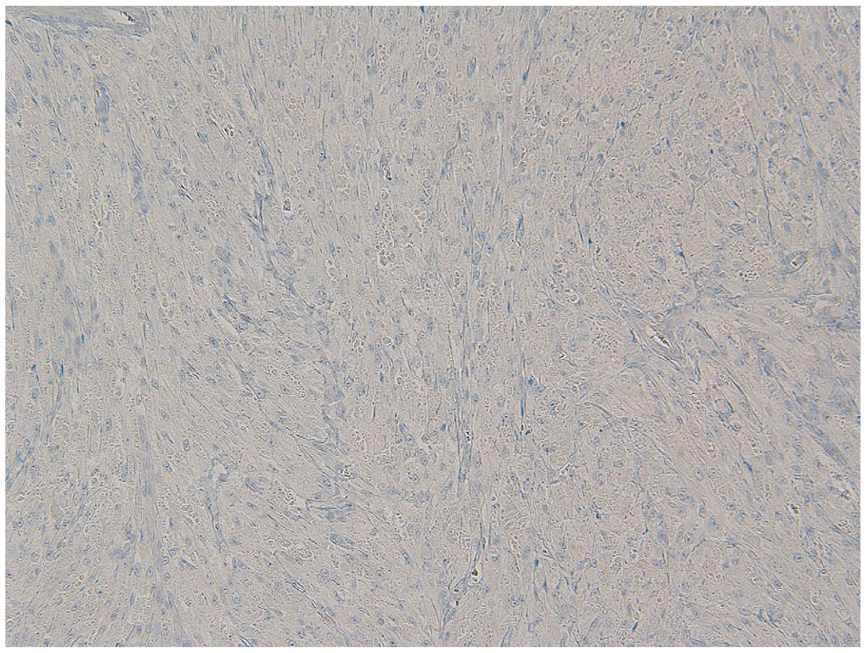
SOX10 is negative in granular cells.

NSE is negative in granular cells.
Discussion
Since first reported by LeBoit in the 1990s, non-neural granular cell tumors have been described in patients between 5 and 83 years. In addition to our patient’s young age, our case is unusual as PNGCT shows a female predominance. 4 Another unique clinical feature in our patient was the rapid growth of the lesion. PNGCTs are typically small in size and have a reported range of 0.2 to 2.8 cm (median: 0.5–0.8 cm). 5 While often seen on the trunk, Rawal et al. presented two cases that were in the buccal mucosa and hard palate. 6 Like the skin lesions, the oral lesions have been reported in a wide age range. The tongue, vermilion of the lower lip, hard palate, alveolar ridge over tooth extraction site, and buccal mucosa are the most commonly affected anatomic areas. 6
The tumor typically arises within the dermis, and despite the atypia and frequent mitotic figures, most articles show excision is curative. 3 Further complicating this rare entity is that lymph node metastasis has been reported, but follow-up of these patients showed no residual disease.5,7,8 Although the tumor is usually superficially located, extension beyond the dermis with infiltration into adjacent eccrine coils has been seen. 9 Even with invasive tumor and lymph node metastasis, the role of lymph node dissection or sentinel biopsy in PNGCT is not currently clear.
Histologically, there is no distinction between a PNGCT that will metastasize versus those that will not. 9 Postoperative follow-up of our patient at 44 months showed no evidence of lymphadenopathy or reoccurrence of the tumor. The clinical features and follow-up of the 13 pediatric patients are summarized in Table 1.
Characteristics of cutaneous non-neural granular cell tumors in children.

N/A indicates that result was not available.
In addition to the atypia typically present in PNGCT, these tumors lack the pseudoepitheliomatous hyperplasia that is so common to conventional granular cell tumors. 5
Furthermore, non-neural granular cell tumors are usually well-circumscribed lesions with vesicular nuclei, pleomorphism, and averaging 5-6/10 HPF (high-power field) mitosis, while conventional granular cell tumors typically have poorly circumscribed margins and no mitotic figures. 11 In addition, the location of the tumor varies, with conventional granular cell tumors most often found in the tongue and gastrointestinal sites, while PNGCT is typically found on the trunk in the superficial dermis.
Ultrastructurally, PNGCTs, like conventional granular cell tumors, show primitive cells with secondary lysosomes of varying sizes. The lysosomal granules in electron microscopy are indistinguishable from those seen in the conventional variant. 5 The tumors do not show clear evidence of neural differentiation with absent S100 protein and NSE. 5 Congenital (gingival) granular cell tumor, which occurs exclusively on the alveolar ridge of the mandible and maxilla of infants, is S100 negative as well. Little is currently known of these rare oral tumors or their relationship to PNGCT. At the date of this review, there is no clear cell of origin for PNGCT; however, a recent paper has suggested that this tumor may arise from the hair follicle and represent a granular cell dermal root sheath fibroma. 10 The granular cells are usually positive for NKI-C3 and CD68, both stains reflecting a non-specific reaction to lysosomes. 11
There are some tumors that they have granular changes due to lysosome accumulation and remain important considerations in patients with PNGCT. These include conventional granular cell tumor, smooth muscle neoplasms, dermatofibromas, epithelioid cell histiocytomas, dermatofibrosarcoma protuberans, fibrous papules, atypical fibroxanthomas, perineuromas, basal cell carcinomas, and metastatic carcinomas. 9 PNGCT is negative for desmin, cytokeratin, Smooth muscle actin SMA, and CD34, effectively ruling out most of the differential diagnosis. 4 Another diagnostic consideration includes melanoma or a benign melanocytic lesion showing granular cell change. Once again, immunohistochemistry separates these lesions from PNGCT as melanocytic lesions are positive for S100 protein and additional melanocytic markers such as HMB-45, MART-1, or MITF. 5
Conclusion
PNGCT is a rare tumor with indolent behavior despite its potential to metastasize. Recognition of this tumor in pediatric patients is essential to avoid overtreatment and incorrect interpretation of the atypia and mitotic activity often present. While metastasis to lymph nodes has been reported in some cases, the indication of sentinel lymph node biopsy is not clear. This unique tumor needs additional studies to further understand the etiology.
Footnotes
Authors’ note
This study was presented as a poster at the College of American Pathologists 2017 Annual Meeting (CAP17), National Harbor, Maryland.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent
Written informed consent was obtained from a legally authorized representative(s) for anonymized patient information to be published in this article.