
Abstract
Objective
Colorectal cancer (CRC) patients with high microsatellite instability (MSI-H) and mismatch repair deficiency (dMMR) had heterogeneous pathology and distinct prognoses. This study aimed to examine the difference in the gene expression profile of dMMR/MSI-H CRC patients with different disease stages and explore the different molecular mechanisms of disease progression.
Methods
A total of 47 patients with dMMR/MSI-H CRC were enrolled and retrospectively studied, including 27 stage II and 20 stage IV patients. Each patient had paired tumor tissue and white blood cell samples, which were analyzed by next-generation sequencing (NGS) of 416 cancer-relevant genes. Pathway enrichment analysis was then performed to analyze the disease stage-specific signaling pathways.
Results
A total of 2878 mutation sites, spanning 378 mutated genes, were detected from the 47 dMMR/MSI-H CRC patients. The mutation frequencies of SMARCA4, EPHA3, MTHFR, RAD50, and PDGFRB were significantly higher in stage II patients than in stage IV patients (p < 0.05), whereas the stage II patients had significantly lower mutation frequencies of TSC2, FGFR1, PTPN13, SMAD3, and STK11 than stage IV patients (p < 0.05). Sixty-three mutated genes were unique to stage II tumors, while 36 mutated genes were exclusively present in stage IV tumors. Pathway analyses demonstrated the PI3K-AKT pathway was shared by both stage II and stage IV tumors, whereas multiple other signaling pathways showed disease stage-specific enrichment.
Conclusion
There were profound differences in mutational profile and molecular mechanisms between stage II and stage IV dMMR/MSI-H CRC.
Introduction
Colorectal cancer (CRC) is one of the most common and fast-growing cancer types worldwide. Based on GLOBOCAN 2020 estimates, globally, there were 1.15 million new colon cancer cases, 0.7 million new rectal cancer cases in 2020. As progress continues, these figures are expected to rise to 1.92 million, 1.16 million, and 78,000, respectively, by 2040.1,2 CRC is well acknowledged to be a heterogeneous disease with various underlying molecular mechanisms. The pathological complexity of CRC is mainly due to its genome instability, leading to the further accumulation of mutations in oncogenes and tumor suppressors. Genome instability includes chromosomal instability and microsatellite instability (MSI).3,4 High MSI (MSI-H) was closely associated with mismatch repair deficiency (dMMR) in CRC and was found in 15%–20% of stage II and III CRCs. dMMR/MSI-H CRC patients tended to have a better prognosis than those with stable microsatellite and proficient mismatch repair (pMMR) tumors 5 ; however, dMMR/MSI-H CRC patients who suffered from recurrence or metastasis could have a poor prognosis due to their insensitivity to chemotherapy. 6 The disease stage also played an important role in determining the prognosis of dMMR/MSI-H CRC patients. 7
Given the heterogeneity of dMMR/MSI-H CRC and the varying responses to treatments across different stages, identifying stage-specific genetic alterations and pathways is crucial for developing targeted therapies and improving patient outcomes. This study aims to fill this gap by comparing the mutational profiles and molecular mechanisms between stage II and stage IV dMMR/MSI-H CRC, with the potential to inform clinical decision-making and therapeutic strategies.
Materials and methods
Study participants
Forty-seven CRC patients who were treated in the Chinese People's Liberation Army (PLA) General Hospital (Beijing, China) from January 2016 to December 2019 were included in this study. The inclusion criteria were (1) patients with pathologically confirmed primary CRC; (2) high-throughput sequencing was carried out within three months of obtaining the tumor tissue samples; (3) patients who were classified as dMMR/MSI-H by the sequencing results; and (4) patients who consented to participate in this study. Tumor tissue samples were obtained via fine-needle aspiration (FNA) from primary tumor sites under ultrasound or CT guidance. All 47 patients had paired formalin-fixed, paraffin-embedded (FFPE) tumor tissue and white blood cell (WBC) samples available for sequencing analysis. All tumor tissue samples were obtained from the primary tumor sites. Written informed consent was required for every patient before we tested his/her DNA samples for research purposes. Patients’ demographic and clinical characteristics were obtained from electronic medical records and telephone interviews. This study was approved by the Institutional Ethics Committees of the Chinese PLA General Hospital (location: Beijing; approval number: S2017-009-01; and date of approval: 2017-2-23).
This study was conducted in accordance with the Declaration of Helsinki (1975, as revised in 2024) and Good Clinical Practice guidelines. The protocol and its amendments were approved by the institutional review board. All patients provided written informed consent.
Lynch syndrome exclusion workflow
All patients with dMMR/MSI-H tumors underwent immunohistochemistry (IHC) for mismatch repair (MMR) proteins (MLH1, MSH2, MSH6, and PMS2). Cases with loss of MLH1/PMS2 were further tested for MLH1 promoter methylation and BRAF V600E mutation. Patients with confirmed MLH1 promoter methylation or BRAF V600E mutation were considered sporadic CRC and retained in the study.
Patients with loss of MSH2/MSH6 or isolated loss of PMS2 or MSH6, as well as those with suspected Lynch syndrome based on IHC or family history, underwent germline testing for pathogenic variants in MLH1, MSH2, MSH6, PMS2, and EPCAM. Individuals with confirmed germline mutations associated with Lynch syndrome were excluded from the study.
High-throughput sequencing and data analysis
According to the manufacturer's instructions, samples were loaded on the Illumina HiSeq 4000 high-throughput sequencing platform (Illumina, Inc., San Diego, CA, USA, https://www.illumina.com). The sequencing panel covered 416 cancer-related genes (Nanjing Geneseeq Technology, Inc., Nanjing, China), spanning 1.46 megabases (Mb) of the human genome. DNAs formed DNA clusters on the flow cell in the assay kit. High-throughput sequencing was carried out on the sequencing platform through cycles of single-base synthesis and interruption, fluorescence detection, and resumption of synthesis. High-throughput sequencing results were aligned with the Chinese hg19 genome data, and gene mapping was completed. The mapping results simultaneously analyzed point mutations, indel mutations, copy number variations, and chromosome structural abnormalities. The 1000 Genomes Project database and database of single-nucleotide polymorphisms (dbSNP) were used for alignment to analyze tumor-specific mutations and germline mutations. We grouped variants with a population frequency of <1% into concomitant SNPs based on the 1000 Genome Project (https://www.internationalgenome.org/) or the 65,000-exome database (http://exac.broadinstitute.org) of the Exome Aggregation Consortium and excluded them from further analysis.
Patient survival staging data and survival data, pathway enrichment, and gene mutation frequency analyses
Venn diagrams are used to analyze differentially expressed genes. The tool used is Venny2.0 (http://bioinfogp.cnb.csic.es/tools/venny/index.html). Gene Ontology (GO)8 functional annotation was performed using the annotation, visualization, and ensemble discovery database (DAVID), and Kyoto Encyclopedia of Genes and Genomes (KEGG) 9 pathway enrichment analysis of the observed set of mutated genes was completed using the KEGG database to obtain the biological functions and possible signaling pathways involved in differentially expressed genes. Particularly, these bioinformatics tools can be used to identify pathway enrichment for tumor-related gene mutations. 10 The screening criteria are p < 0.05. Gene mutation frequency analysis was performed according to the literature. 11 In addition, we obtained the clinical data and expression profiles of CRC patients from the TCGA database and performed prognostic correlation analysis of the observed genes.
Gene set enrichment analysis (GSEA)
GSEA is a computational method used to determine whether a priori-defined gene sets show statistically significant consistent differences between two biological states. We ranked the genes obtained at stage II and stage IV, respectively. GSEA was used to analyze the KEGG and GO pathways for both sets of gene sets. The calculation yielded an enrichment score (ES).
Statistical analysis
The SPSS 23.0 statistical software was used for all the analyses. Qualitative data were expressed as percentages, and the chi-square test was used for statistical analysis. The Kaplan-Meier curve was used for univariate survival analysis. A difference with a p-value < 0.05 was considered to be statistically significant.
Results
Patient demographic and clinical characteristics
A total of 47 patients with dMMR/MSI-H CRC were enrolled in our study, of which 27 and 20 had stage II and stage IV disease, respectively. Table 1 shows the baseline information of the two groups. Among the 47 patients, the median age was 48 years, ranging from 26 to 80 years. Thirty-one patients (66.0%) were males, and 16 patients (34.0%) were females. Nineteen (70.4%) of the stage II patients and 12 (60.0%) of the stage IV patients had a family history of CRC. In addition, 1, 21, and 25 patients had lesions in the bilateral colon, right colon, and left colon and rectum, respectively. Twenty-nine patients were diagnosed with adenocarcinoma, while 18 patients had mucinous adenocarcinoma.
General information on the included patients.

Overall mutational profile
A total of 2878 somatic mutation sites, spanning 378 genes, were found in the tumors of the 47 patients. The top five mutated genes were APC (68.09%), KMT2B (61.70%), TGFBR2 (61.70%), ARID1A (57.45%), and NOTCH1 (53.19%), and most of these frequently mutated genes were shared between stages II and IV patients (Figure 1(a) and (b)). Among these identified genetic alterations, 84.68% were single-nucleotide missense mutations, 23.28% were frameshift mutations, 6.39% were nonsense mutations, and 0.5% were gene rearrangements.

The frequently mutated genes detected in dMMR/MSI-H CRC patients. (a) The top 30 frequently mutated genes in stage II dMMR/MSI-H CRC patients. (b) The top 30 frequently mutated genes in stage IV dMMR/MSI-H CRC patients. dMMR: mismatch repair deficiency; MSI-H: high microsatellite instability; CRC: colorectal cancer.
Identification of enriched mutations for different disease stages
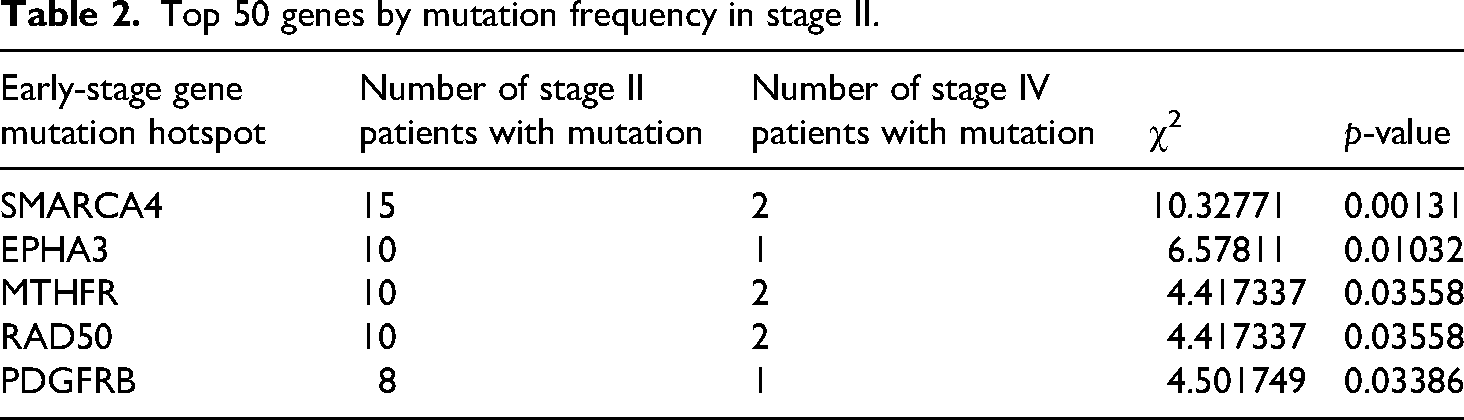
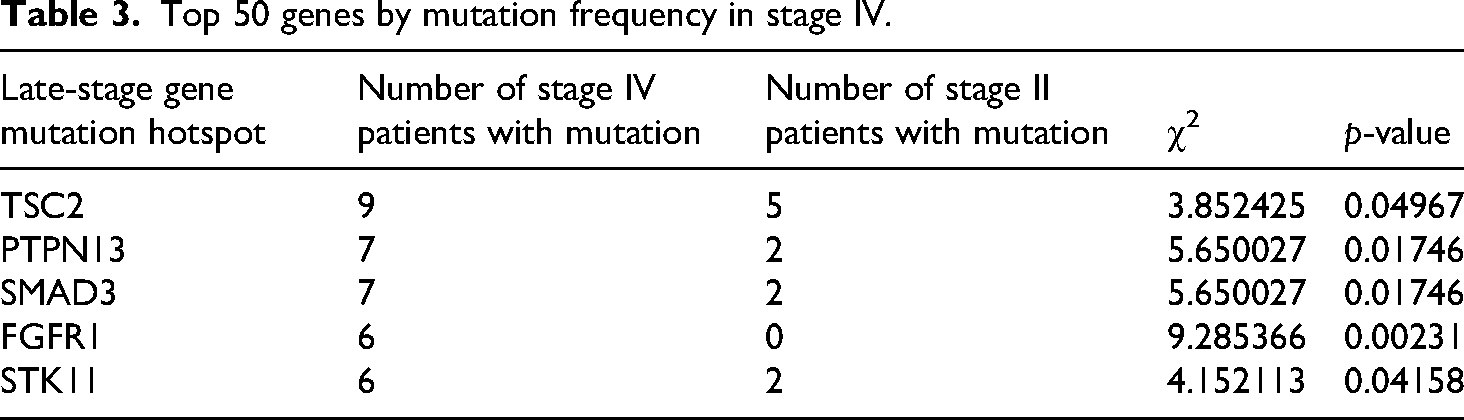
By analyzing the mutation frequency of the top 50 genes in patients with different disease stages, we found that SMARCA4 (p = 0.00131), EPHA3 (p = 0.01032), MTHFR (p = 0.03558), RAD50 (p = 0.03558), and PDGFRB (p = 0.03386) mutations were significantly enriched in stage II dMMR/MSI-H CRC rather than stage IV dMMR/MSI-H CRC (Table 2). In contrast, multiple genes had significantly higher mutation rates in stage IV patients than in stage II patients, including TSC2 (p = 0.04967), FGFR1 (p = 0.00231), PTPN13 (p = 0.01746), SMAD3 (p = 0.01746), and STK11 (p = 0.04158) (Table 3).
Top 50 genes by mutation frequency in stage II.
Top 50 genes by mutation frequency in stage IV.
Pathway enrichment analysis for different disease stages
We then performed the pathway analysis for all the mutations detected in either stage II or stage IV patients. As shown in Figure 2, one of the most outstanding oncogenic pathways detected in stage II dMMR/MSI-H CRC was the PI3K-AKT signaling pathway. On the other hand, for stage IV dMMR/MSI-H CRC, we detected several additional oncogenic pathways besides PI3K-AKT, such as the RAS-MAPK signaling pathways.

Pathway enrichment for different disease stages. (a) Pathway enrichment analysis using all the detected mutations in stage II dMMR/MSI-H CRC patients. (b) Pathway enrichment analysis using all the detected mutations in stage IV dMMR/MSI-H CRC patients. dMMR: mismatch repair deficiency; MSI-H: high microsatellite instability; CRC: colorectal cancer.
GSEA analysis results
In the two-stage GSEA analysis, pathways associated with tumorigenesis showed significant enrichment. In Stage II, significant enrichment was shown in both the Notch signaling pathway (ES = 0.6906 and NP = 0.0221) and the basal cell carcinoma pathway (ES = 0.8080 and NP = 0.0000) (Figure 3). The role of the Notch signaling pathway as an oncogene or tumor suppressor in a variety of cellular environments has received extensive attention, and its dysregulation plays an extremely important role in the occurrence and progression of human hematologic malignancies and solid tumors. In addition, the significant enrichment of the basal cell carcinoma pathway also highlights the potential importance of this pathway in tumorigenesis.

GSEA performed by the KEGG database and the GO database using all mutations detected in patients with stage II dMMR/MSI-H CRC. GSEA: gene set enrichment analysis; KEGG: Kyoto Encyclopedia of Genes and Genomes; GO: Gene Ontology; dMMR: mismatch repair deficiency; MSI-H: high microsatellite instability; CRC: colorectal cancer.
In Stage IV, the basal cell carcinoma pathway (ES = 0.6408 and NP = 0.0253) and Notch signaling pathway (ES = 0.6663 and NP = 0.0227) continued to show significant enrichment, which is consistent with Stage II findings and further supports the continued role of this pathway in tumor progression. In addition, the mismatch repair pathway (ES = 0.6832 and NP = 0.0273) showed significant significance, which supported the close association between gene mismatch repair and tumor development. Other pathways directly associated with tumors, such as lactate metabolism (ES = 0.6343 and NP = 0.0722), also showed significant enrichment, which may be related to metabolic reprogramming of tumor cells (Figure 4). These findings highlight that the enrichment patterns of specific pathways may be closely related to the biology and progression stage of the tumor at different stages of tumor development.

GSEA performed by the KEGG database and the GO database using all mutations detected in patients with stage IV dMMR/MSI-H CRC. GSEA: gene set enrichment analysis; KEGG: Kyoto Encyclopedia of Genes and Genomes; GO: Gene Ontology; dMMR: mismatch repair deficiency; MSI-H: high microsatellite instability; CRC: colorectal cancer.
Pathway enrichment analysis for stage-specific mutations
Among the 378 mutated genes, 279 were present in stage II and stage IV dMMR/MSI-H CRC. A total of 63 and 36 mutated genes were specific to stage II and stage IV tumors, respectively (Table 4 and Figure 5). We, thereby, analyzed the differentially enriched signaling pathways using the mutations specific to either stage II or stage IV tumors. As shown in Figure 6, there were generally more signaling pathways enriched in stage IV patients than in stage II patients. Particularly, the metabolism-related pathways showed the greatest differences between stage II and stage IV patients in terms of the highest gene ratio and p-value differences. Furthermore, stage IV-specific mutations were highly enriched in multiple pathways, including the HIF-1, Insulin, Rap1, JAK-STAT, and VEGF signaling pathways, whereas stage II-specific mutations were mainly involved in the GnRH and Fanconi anemia signaling pathways (Figure 6).

A Venn diagram formed by all mutations detected in stage II dMMR/MSI-H CRC patients, all mutations detected in stage IV dMMR/MSI-H CRC patients, and genes with prognostic significance among these mutations. dMMR: mismatch repair deficiency; MSI-H: high microsatellite instability; CRC: colorectal cancer.

Pathway enrichment analysis using mutations specific to stage II and stage IV dMMR/MSI-H CRC. dMMR: mismatch repair deficiency; MSI-H: high microsatellite instability; CRC: colorectal cancer.
Gene mutations specific to different stages.

Prognosis-related genes and their prognostic significance for CRC
We used the patient survival data of stage II and stage IV in the TCGA database to screen for genes that are associated with prognosis among all mutated genes. In this analysis, we assessed the relationship between the genes associated with the hazard ratio (HR) and found that all statistically significant genes had HRs > 1, indicating that they were associated with higher risk (Figure 7). Specifically, there were 10 statistically significant genes, which were AIP (HR = 1.50, 95% CI: 1.01–2.23, p = 0.04), CDKN2A (HR = 1.19, 95% CI: 1.04–1.35, p = 0.01), CUX1 (HR = 1.43, 95% CI: 1.00–2.06, p = 0.04), FLT1 (HR = 1.29, 95% CI: 1.01–1.63, p = 0.03), FLT4 (HR = 1.25, 95% CI:1.01–1.53, p = 0.04), GSTM1 (HR = 1.09, 95% CI:1.01–1.19, p = 0.03), MEF2B (HR = 1.42, 95% CI:1.12–1.81, p = 2.8 × 10−3), NOTCH3 (HR = 1.23, 95% CI:1.01–1.51, p = 0.04), SRY (HR = 1.68, 95% CI: 1.13–2.50, p = 0.04), and XPA (HR = 1.59, 95% CI: 1.05–2.41, p = 0.03). Of these 10 salient genes, nine occur in mutated genes common to stage II and stage IV patients, and one comes from a mutated gene unique to stage IV patients (Figure 5). Further, we used the survival data and expression profiles of all CRC patients in the TCGA database to plot the Kaplan-Meier curve of these 10 genes (Figure 8).
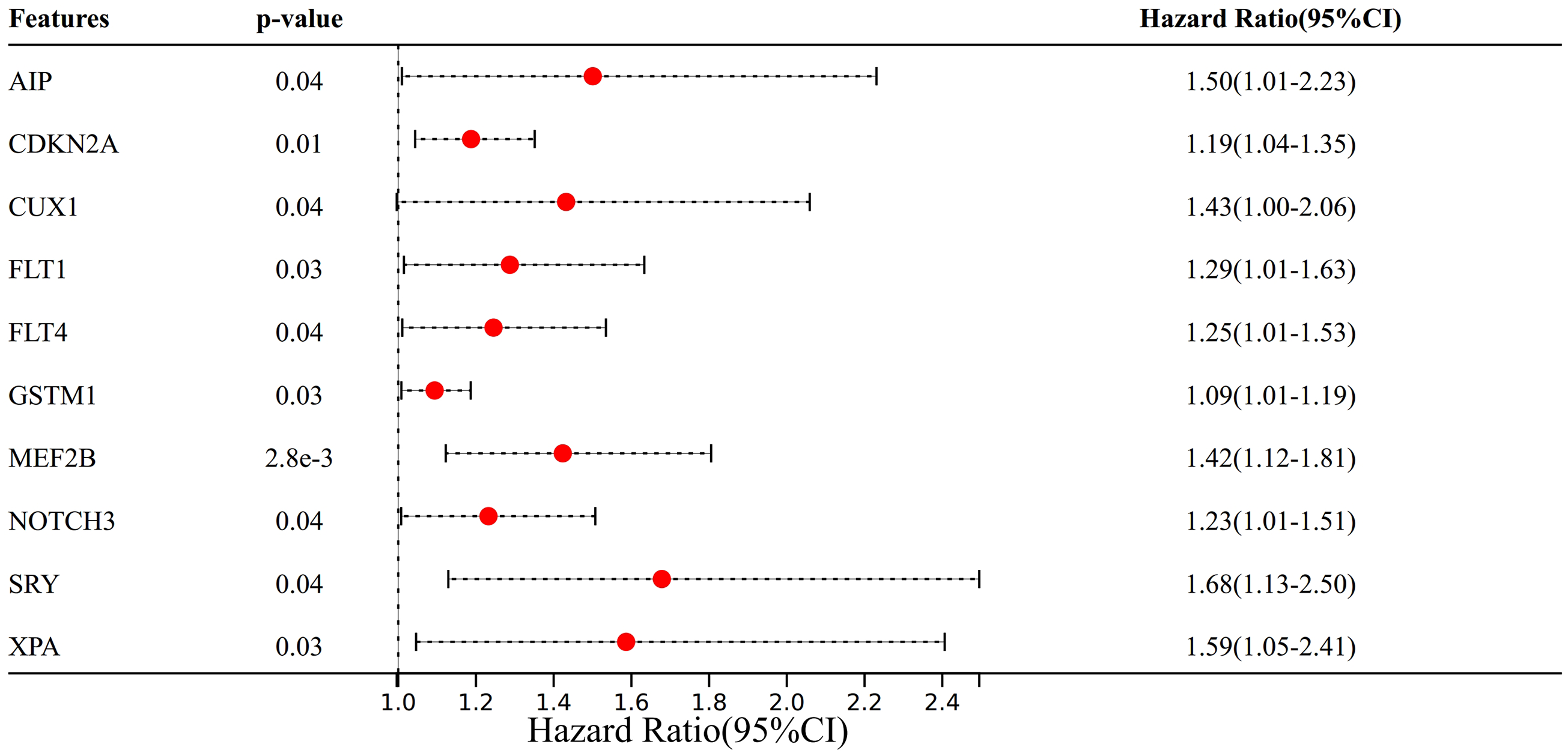
Forest plot illustrating the association between various genetic features and HRs in stage II and IV dMMR/MSI-H CRC patients. Each feature is represented by a red dot indicating the point estimate of the HR, with the horizontal black dashed line representing the 95% CI. The vertical dashed line at HR = 1.0 serves as a reference for no effect. Features with p-values < 0.05 are highlighted, suggesting statistical significance. HRs: hazard ratios; dMMR: mismatch repair deficiency; MSI-H: high microsatellite instability; CRC: colorectal cancer; CI: confidence interval.
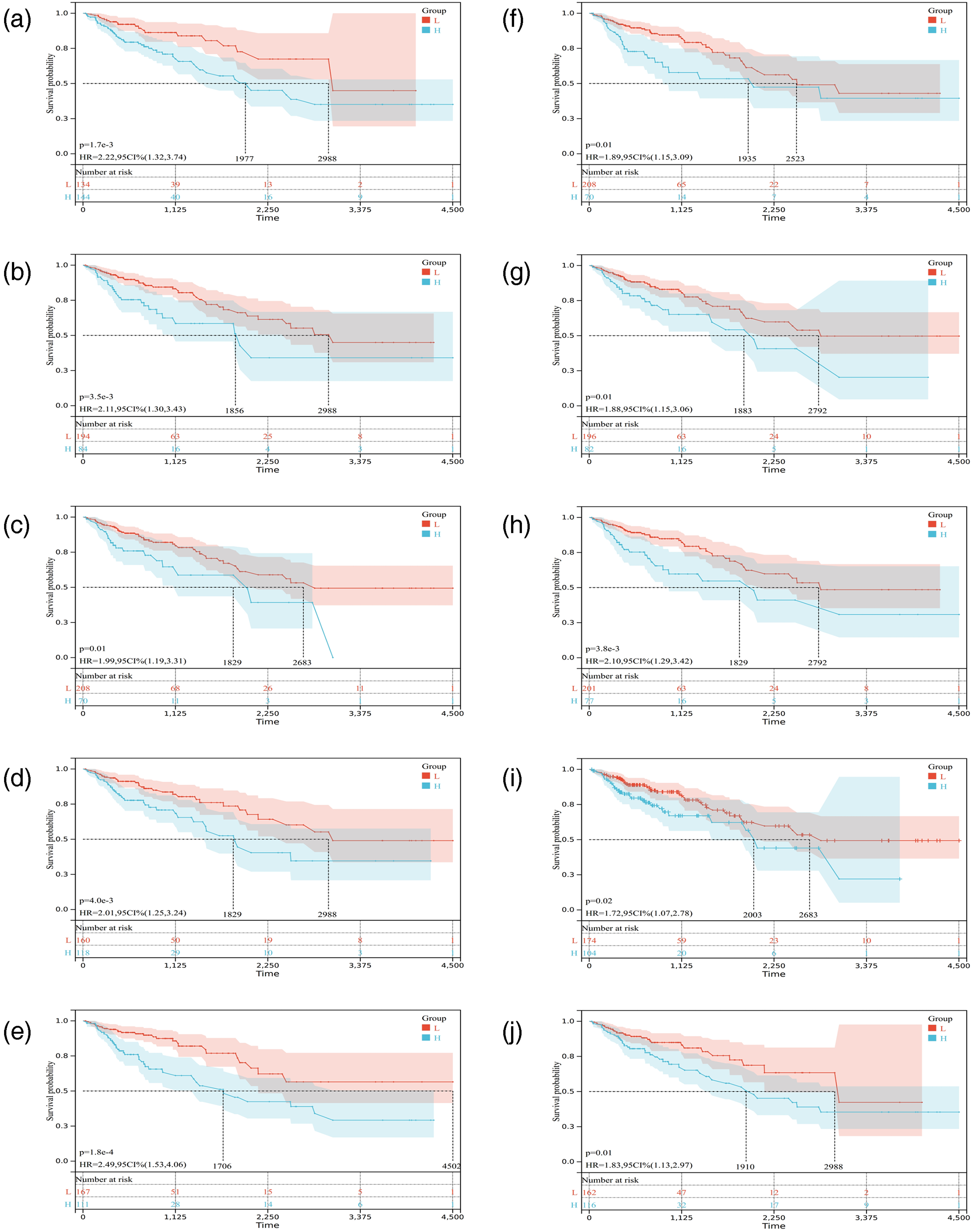
Kaplan-Meier survival curves for ten genetic features associated with the prognosis of patients. (a) Correlation between AIP and prognosis in patients with CRC, log-rank p = 1.7 × 10−3, HR = 2.22, 95% CI: 1.32–3.74. (b) Correlation between CDKN2A and prognosis in patients with CRC, log-rank p = 3.5 × 10−3, HR = 2.11, 95% CI: 1.30–3.43. (c) Correlation between CUX1 and prognosis in patients with CRC, log-rank p = 0.01, HR = 1.99, 95% CI: 1.19–3.31. (d) Correlation between FTL1 and prognosis in patients with CRC, log-rank p = 4.0 × 10−3, HR = 2.01, 95% CI: 1.25–3.24. (e) Correlation between FTL4 and prognosis in patients with CRC, log-rank p = 1.8 × 10−4, HR = 2.49, 95% CI: 1.53–4.06. (f) Correlation between GSTM1 and prognosis in patients with CRC, log-rank p = 0.01, HR = 1.89, 95% CI: 1.15–3.09. (g) Correlation between MEF2B and prognosis in patients with CRC, log-rank p = 0.01, HR = 1.88, 95% CI: 1.15–3.06. (h) Correlation between NOTCH3 and prognosis in patients with CRC, log-rank p = 3.8 × 10−3, HR = 2.10, 95% CI: 1.29–3.42. (i) Correlation between SRY and prognosis in patients with CRC, log-rank p = 0.02, HR = 1.72, 95% CI: 1.07–2.78. (j) Correlation between XPA and prognosis in patients with CRC, log-rank p = 0.01, HR = 1.83, 95% CI: 1.13–2.97. HR: hazard ratio; CRC: colorectal cancer; CI: confidence interval.
Discussion
In this study, we compared the differences in the gene mutation spectrum between stage II and stage IV dMMR/MSI-H CRC. To the best of our knowledge, our study was the first to examine the disease stage-based differential mutational profiles and molecular mechanisms in Chinese dMMR/MSI-H CRC patients, and our findings helped better understand the genetic basis of the disease progression in dMMR/MSI-H CRC.
Our study reveals distinct mutational profiles and molecular mechanisms between stage II and stage IV dMMR/MSI-H CRC, which have significant clinical implications. The differential enrichment of mutations in genes like TSC2, FGFR1, and SMAD3 in stage IV patients suggests that targeting these pathways could be beneficial in advanced stages. Additionally, the identification of prognostic genes such as AIP, CDKN2A, and CUX1 provides potential biomarkers for risk stratification and treatment planning. These findings underscore the importance of personalized treatment approaches based on disease stage and genetic profile.
Although 31 patients reported a family history of CRC, comprehensive IHC screening plus MLH1 promoter methylation/BRAF V600E testing and germline analysis confirmed that none fulfilled the genetic criteria for Lynch syndrome. In the mutation landscape analysis, we employed high-throughput sequencing and found that the mutation frequency of SMARCA4, EPHA3, MTHFR, RAD50, and PDGFRB was significantly higher in stage II patients than in stage IV patients. On the other hand, the mutation frequency of TSC2, FGFR1, PTPN13, SMAD3, and STK11 in stage IV patients was significantly higher than those in stage II patients. SMARCA4, PDGFRB, and EPHA3 were well-known upstream effector proteins for multiple signaling pathways,12–14 while RAD5 and MTHFR were involved in DNA damage repair and methylation.15,16 Among the hotspot mutations in stage IV patients, TSC2, SMAD3, FGFR1, and PTPN13 were effector proteins of major signaling pathways, such as the PI3K-AKT and transforming growth factor β (TGF-β) signaling pathways,17–20 and mutations in these genes tended to cause dysregulated cell division, proliferation, and migration, leading to the uncontrolled tumor growth.21–24 Besides, GSEA analysis revealed significant enrichment of pathways associated with tumorigenesis, providing valuable insights into the molecular underpinnings of CRC progression. In stage II, we observed notable enrichment in the Notch signaling pathway and the basal cell carcinoma pathway. The Notch signaling pathway, known for its dual role as an oncogene or tumor suppressor, has been implicated in the development and progression of various cancers, including hematologic malignancies and solid tumors. Its dysregulation is crucial in the oncogenic process, and our findings underscore its relevance in early-stage CRC. The basal cell carcinoma pathway, which also showed significant enrichment, suggests a potential role in the early stages of tumorigenesis, which warrants further investigation. In stage IV, the continued significant enrichment of both the basal cell carcinoma and Notch signaling pathways aligns with our stage II findings, reinforcing their importance throughout CRC progression. The emergence of the mismatch repair pathway as significantly enriched in stage IV is consistent with the established link between gene mismatch repair and tumorigenesis. Additionally, the enrichment of the lactate metabolism pathway hints at the metabolic reprogramming of tumor cells, which is a recognized hallmark of cancer. These findings suggest that the enrichment patterns of specific pathways are intricately linked to the biological characteristics and progression stages of CRC, highlighting the dynamic nature of the disease.
We discovered several potential differences in molecular mechanisms between stage II and stage IV dMMR/MSI-H CRC. Stage IV-specific mutations were mainly enriched for tumor growth, proliferation, and malignant transformation-related pathways, such as RAP1, JAK-STAT, and VEGF signaling pathways.25–27 RAP1, which contains RAP1A and RAP1B isoforms, is a member of the RAS small G protein family, 28 and RAP1 deletion could disrupt cell–cell adhesion and promote the invasiveness of cancer cells.29,30 Other studies reported that RAP1B overexpression activated the PI3K/AKT/mTOR signaling pathway in gastric cancer and was a poor prognostic factor.31,32 In addition, the JAK-STAT signaling pathway was associated with immune responses, inflammation, and cell proliferation,33,34 and aberrant activation of STAT3 in colon cancer was associated with poor prognosis. 35 Morikawa et al. 36 analyzed STAT3 activation in 725 colon cancer patients and found that STAT3 tyrosine phosphorylation levels were significantly elevated in late-stage cancer tissues and lymph node metastases. Activation of STAT3 in tumor cells could also result in aberrant promoter methylation or protein tyrosine phosphatase inactivation, leading to decreased expression of suppressor of cytokine signaling (SOCS) proteins and persistent STAT3 activation. 37 The VEGF signaling pathway was associated with angiogenesis, and its disruption played an irreplaceable role in tumorigenesis and metastasis. 27 At present, antiangiogenic drugs have become promising treatments for CRC. Overall, stage II and stage IV dMMR/MSI-H CRC exhibited significantly different mutation spectra and molecular mechanisms, which may at least partially explain the dramatic difference in treatment outcomes between the two groups of patients.
In dMMR/MSI-H tumor tissues, aberrant DNA mismatch repair results in accumulated gene mutations, thereby leading to a large number of neoantigens. The immune status of early stage patients is usually at a normal level without suppression of lymphocyte activity. In contrast, the late-stage patients tended to have an aberrant PD-L1 expression and immune evasion,38,39 resulting in impaired ability to recognize and clear tumors. Our study showed that the PI3K-AKT and the RAS-MAPK were the most pronounced signaling pathways that were mutated in stage IV patients, and previous studies have shown that mutations in these pathways could suppress the immune system.40–45 In addition, we found that IL7R was exclusively mutated in stage IV tumors. IL-7R mediated several signaling pathways that were vital for the development and homeostasis of normal T cells.28,46 Therefore, IL7R mutations could potentially affect immunity during disease progression and might play a role in determining the sensitivity to PD-1 treatments.
Our analysis of the TCGA database identified ten genes with statistically significant HRs > 1, indicating their association with a higher risk of poor prognosis. These genes include AIP, CDKN2A, CUX1, FLT1, FLT4, GSTM1, MEF2B, NOTCH3, SRY, and XPA. The presence of nine of these genes in mutated genes common to both stage II and stage IV patients, and one unique to stage IV, suggests a shared genetic landscape that contributes to CRC progression, with additional mutations specific to advanced stages. The Kaplan-Meier curves for these genes, constructed using survival and expression data from all CRC patients in the TCGA database, further elucidate their prognostic significance.
The identification of these genes offers a foundation for understanding the genetic determinants of CRC prognosis and may inform the development of targeted therapies. The overlap between stages suggests that while certain genetic alterations are common across the disease spectrum, others are specific to advanced stages, potentially influencing treatment response and survival. Treatment details for all patients with available data are summarized in Supplemental Table 1.
Our study has several limitations. Firstly, the analysis was confined to stages II and IV of CRC, which limited our understanding of the disease's progression across all stages. Future studies should consider including more stages to fully elucidate the molecular changes in CRC. Secondly, the limited sample size affected the depth of our statistical analysis; subsequent studies should expand the sample size and consider validating results using data from public databases. Besides, the tumor mutation burden (TMB) data were not available for all patients due to the relatively short follow-up period at the time of analysis. Although we have provided TMB values for a subset of patients in the Supplemental Material (Supplemental Table 2), the incomplete dataset may limit the comprehensive evaluation of TMB as a biomarker in this cohort. Although we summarized the treatment regimens received by patients in both stage II and stage IV groups, the relatively short follow-up time and incomplete survival data currently preclude a robust analysis of the association between specific therapies and clinical outcomes. TMB values were available for 12 patients and are provided in Supplemental Table 2; unfortunately, TMB data for the remaining cases could not be retrieved because of the short follow-up period at the time of analysis (Supplemental Table 1). As follow-up continues and data mature, we plan to conduct more detailed prognostic analyses in future studies to better elucidate the impact of treatment on survival in dMMR/MSI-H CRC patients.
Conclusion
This study reveals distinct mutation profiles and molecular mechanisms between stage II and stage IV dMMR/MSI-H CRC in Chinese patients. Stage IV exhibits enrichment of mutations (e.g. TSC2, FGFR1, and SMAD3) and pathways (e.g. RAP1, JAK-STAT, and VEGF) driving proliferation, immune evasion, and metastasis, while stage II shows different patterns. Prognostic genes (e.g. AIP, CDKN2A, and CUX1) and pathway dysregulations (e.g. Notch, basal cell carcinoma, mismatch repair, and lactate metabolism) were identified. These stage-specific genetic and molecular differences sheds light on understanding the molecular trajectory during disease progression and facilitates choosing appropriate treatment regimens in dMMR/MSI-H CRC.
Supplemental Material
sj-pdf-1-sci-10.1177_00368504251412580 - Supplemental material for Comparison of the differentially enriched mutations/pathways between stage II and stage IV dMMR/MSI-H colorectal cancer
Supplemental material, sj-pdf-1-sci-10.1177_00368504251412580 for Comparison of the differentially enriched mutations/pathways between stage II and stage IV dMMR/MSI-H colorectal cancer by Chun Han, Sisi Ye, Juan Li, Qian Qiao, Li Bai and Tingting Zhang in Science Progress
Supplemental Material
sj-pdf-2-sci-10.1177_00368504251412580 - Supplemental material for Comparison of the differentially enriched mutations/pathways between stage II and stage IV dMMR/MSI-H colorectal cancer
Supplemental material, sj-pdf-2-sci-10.1177_00368504251412580 for Comparison of the differentially enriched mutations/pathways between stage II and stage IV dMMR/MSI-H colorectal cancer by Chun Han, Sisi Ye, Juan Li, Qian Qiao, Li Bai and Tingting Zhang in Science Progress
Supplemental Material
sj-docx-3-sci-10.1177_00368504251412580 - Supplemental material for Comparison of the differentially enriched mutations/pathways between stage II and stage IV dMMR/MSI-H colorectal cancer
Supplemental material, sj-docx-3-sci-10.1177_00368504251412580 for Comparison of the differentially enriched mutations/pathways between stage II and stage IV dMMR/MSI-H colorectal cancer by Chun Han, Sisi Ye, Juan Li, Qian Qiao, Li Bai and Tingting Zhang in Science Progress
Footnotes
Acknowledgements
We thank the patients and their families, the investigators, and all team members for participating in this study.
Author contributions
LB took part in conception and design. QQ and JL participated in the data extraction. SSY conducted the data analyses and assisted in the interpretation of the results. CH was involved in the interpretation of the results, and drafting of the manuscript. TTZ participated in the critical revision of the manuscript. All authors approved the final draft for submission.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Key Research and Development (R&D) Plan (2016YFC1303602).
Declaration of conflicting interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Data availability statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. Bulk RNA-seq data and corresponding clinical information of CRC patients were obtained from The Cancer Genome Atlas (TCGA) COAD and READ cohorts (https://www.cancer.gov/ccg/research/genome-sequencing/tcga). The raw sequence data have been deposited in the Genome Sequence Archive in the National Genomics Data Center, China National Center for Bioinformation under accession number HRA015153 (![]() ).
).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.