
Abstract
Von Hippel-Lindau (VHL) disease is a rare inherited tumor syndrome characterized by the development of multiple neoplasms. The broad variability in clinical manifestations makes this entity susceptible to being overlooked during clinicpathological diagnosis. A female patient in her late 20s presented with multiple adrenal masses during a routine health check-up, with associated dizziness and palpitations lasting 1 month. Laboratory studies revealed plasma-free normetanephrine level of 2894.9 pg/ml. Abdominal contrast-enhanced computed tomography showed multiple lesions consisting of bilateral adrenal masses and retroperitoneal nodules. Postoperative pathological examination demonstrated the diagnosis of bilateral adrenal pheochromocytomas, retroperitoneal paraganglioma, and pancreatic neuroendocrine tumor. Molecular genetic testing detected a pathogenic germline mutation in the VHL gene (c.499C > T: p.R167 W). Subsequent brain magnetic resonance imaging revealed a hypervascular cerebellar nodule. Genetic and clinical findings confirmed a definitive diagnosis of VHL syndrome type 2B. The diverse manifestations of VHL syndrome often cause diagnostic delays. Analyzing this case alongside the literature highlights the need to suspect VHL in young patients with multiple tumors, for whom genetic testing is crucial for definitive diagnosis. While a single case cannot capture the full disease spectrum, it provides valuable clinical insight.
Introduction
Von Hippel-Lindau (VHL) disease is a rare inherited tumor syndrome characterized by the development of multiple benign and malignant neoplasms. Cardinal manifestations include retinal hemangioblastoma (RH), central nervous system hemangioblastoma (CNS-H), clear cell renal cell carcinoma (ccRCC), pheochromocytoma (PCC) and paraganglioma (PGL), endolymphatic sac tumor, pancreatic neuroendocrine tumor (PNET), renal and pancreatic cyst, and broad ligament and epididymal cystadenoma. 1 Other rare tumors, such as multiple lung adenocarcinoma, have also been reported. 2 This heterogeneity in clinical presentation makes the rare disease prone to oversight. Herein, we report a young female patient whose predominant manifestation was multiple neuroendocrine tumors. A stepwise diagnostic workup ultimately identified VHL syndrome as the underlying etiology, and we conducted a review of the literature to improve identification of this rare entity. It is worth noting that a preliminary version of this case report was previously shared as a preprint. 3
Case report
A female patient in her late 20s was admitted to Gui Qian International Hospital in mid-2020 for multiple adrenal masses during a routine health check-up, with associated dizziness and palpitations lasting 1 month. The patient did not report any medical history of hypertension, diabetes, or coronary heart disease and had no history of prior surgical procedures or injuries. Her father died of an intracranial tumor more than 20 years ago, while her eldest sister passed away a decade ago due to adrenal disease and hypertension during pregnancy (owing to the long time elapsed, family history details for the patients were unobtainable). On admission, the patient's pulse was 72 beats per minute, and blood pressure was 126/89 mmHg. Laboratory studies revealed a markedly elevated plasma-free normetanephrine level of 2894.9 pg/ml (reference range, < 145 pg/ml), while the plasma-free metanephrine level was within the normal range at 41.2 pg/ml (reference range, < 62 pg/ml). Abdominal contrast-enhanced computed tomography (CT) showed multiple lesions consisting of bilateral adrenal masses and retroperitoneal nodules. The left adrenal gland contained two distinct masses with a larger inferior lesion and a smaller superior lesion (Figure 1A). A hypodense mass was observed in the right adrenal gland (Figure 1A). Additional findings included a retro-pancreatic nodule and a para-uncinate mass exhibiting patchy calcifications (Figure 1B). The preliminary clinical diagnosis was multiple PGL/PCCs. The patient maintained stable blood pressure following admission. After preoperative optimization, the patient underwent a combined surgical procedure consisting of laparoscopic left adrenalectomy and open right adrenal surgery with tumor resection, partial adrenalectomy, retroperitoneal tumor resection, and pancreatic tumor excision. Pathological examination demonstrated that the bilateral adrenal masses and the retro-pancreatic nodule were histologically and immunohistochemically similar. All specimens consisted of well-demarcated, grayish-yellow to gray-brown nodular tissue with homogeneous cut surfaces, though the left adrenal masses showed significant cystic degeneration (Figure 2A). The tumor cells were arranged in solid nests surrounded by a dense capillary network (Figure 2B, 2C). Immunohistochemically, they stained positively for chromogranin A (Figure 2D) and synaptophysin but were negative for cytokeratin (Figure 2E). Supporting cells expressed S-100 (Figure 2F). The para-uncinate mass appeared gray-brown with ill-defined borders and a firm, grayish-white cut surface. Neoplastic cells were exhibited in gyrate trabecular and glandular patterns (Figure 3A), exhibiting mild atypia and rare mitoses (approximately 3 mitoses/2 mm²). The stroma ranged from rich capillary networks between tumor nests to broad collagenized and calcified areas. Immunohistochemistry showed positivity for chromogranin A (Figure 3B), synaptophysin, and cytokeratin (Figure 3C), with a Ki-67 labeling index of approximately 5% (Figure 3D). Based on pathological findings, the diagnosis of bilateral adrenal PCCs, retroperitoneal PGL, and PNET G2 were approved. Subsequent genetic testing of peripheral blood samples identified a heterozygous missense mutation in VHL (c.499C > T, p.R167 W), characterized by a cytosine-to-thymine substitution at nucleotide position 499 of the cDNA, resulting in an arginine-to-tryptophan replacement at codon 167. The final diagnosis was VHL syndrome type 2B. The patient's sister and nephew were also screened for a germline VHL mutation, and no pathogenic variant was detected. Concurrently, a contrast-enhanced brain magnetic resonance imaging (MRI) was obtained to assess for potential CNS involvement, identifying a 3.6 mm intensely enhancing nodule in the right cerebellar hemisphere with features suggestive of hemangioblastoma (Figure 1C). In the postoperative phase, the patient was switched from intravenous hydrocortisone (200 mg/day) to oral prednisone (20–30 mg/day) following the resumption of oral intake, with a planned taper to 15–20 mg/day by discharge and further reductions of 2.5 mg every 4 weeks. The normetanephrine level had normalized upon recheck at the time of discharge after the 27-day hospitalization. The patient was advised to undergo regular blood pressure monitoring, plasma metanephrines level checks, and annual imaging surveillance with head MRI and abdominal CT or ultrasound. At the follow-up assessment in mid-2025, the cerebellar nodule was stable in size, and no new abdominal space-occupying lesions were identified. Written informed consent was obtained from the patient for publication. Our team had obtained the consent of patient for treatment. The patient's details have been de-identified. The reporting of this study conforms to CARE guidelines. 4
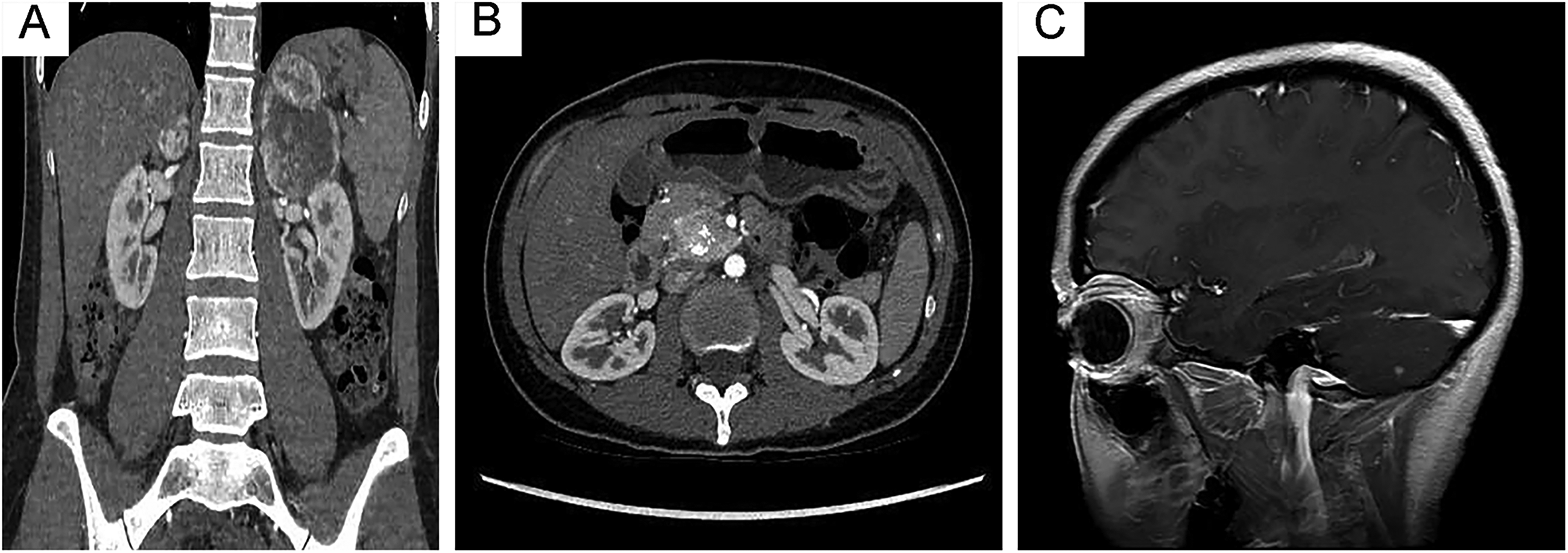
The imaging characteristics of this case. (A) Contrast-enhanced abdominal CT reveals two left adrenal masses: a larger hypodense inferior mass (5.5cm × 4.5cm × 5.2 cm; non-contrast 39 HU, arterial phase 161 HU) and a smaller isodense superior mass (2.2cm × 3.1cm × 2.9 cm; non-contrast 40 HU, arterial phase 201 HU). A hypodense mass (3.5cm × 2.3cm × 1.8 cm; non-contrast 40 HU, arterial phase 201 HU) replaces the right adrenal gland. (B) A para-uncinate mass with patchy calcifications (3.5cm × 2.6cm × 2.8 cm; non-contrast 59 HU, arterial phase 89 HU) is evident. (C) Post-contrast T1-weighted MRI of the brain demonstrates an intensely enhancing 3.6-mm hypervascular nodule in the right cerebellar hemisphere. CT: computed tomography; MRI: magnetic resonance imaging.
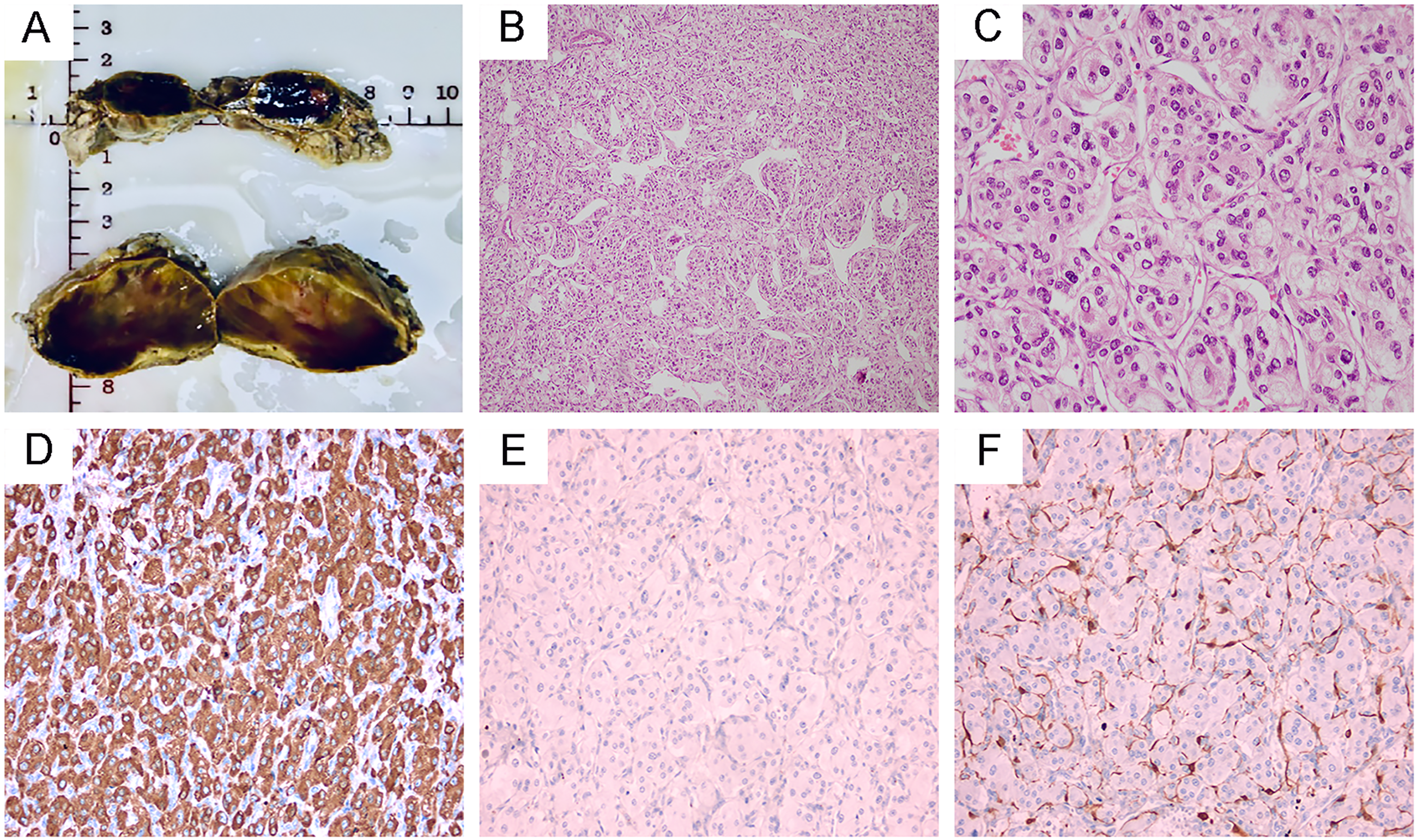
Morphological and immunohistochemical characteristics of the left adrenal masses. Photograph of the left adrenal masses (A). The tumors cells were arranged in solid-nesting (B × 100), surrounded by dense network of capillaries (C × 400). Immunohistochemical studies demonstrated diffuse positive staining for chromogranin A (D × 200) and did not expressed cytokeratin (E × 200). Supporting cells were S-100 positive (F × 200).
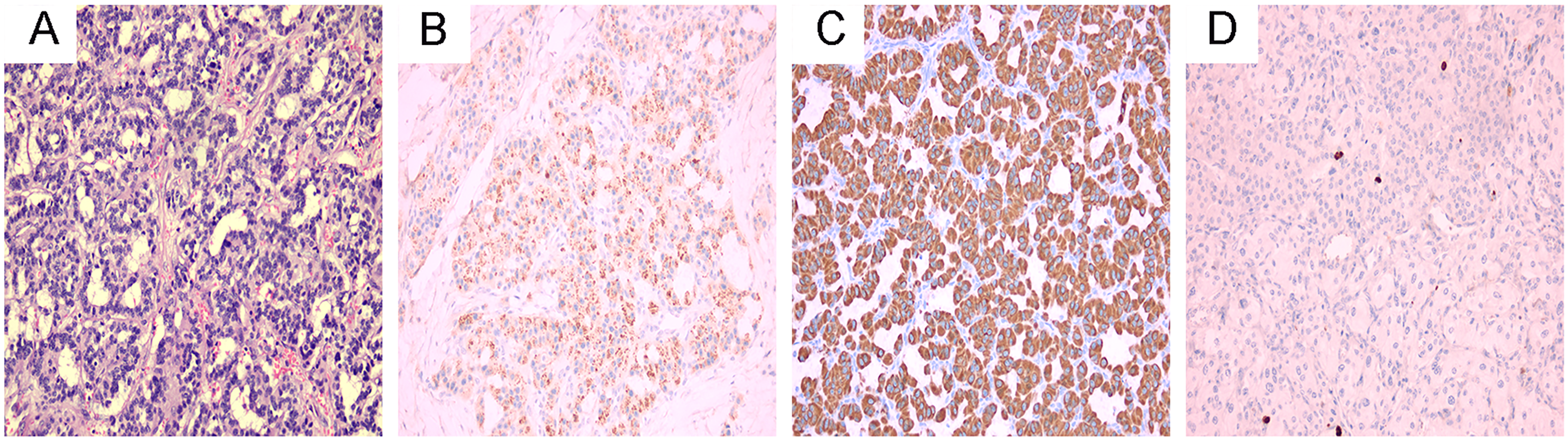
Histological and immunohistochemical features of the para-uncinate mass. The tumor cells arranged in gyriform trabecular and glandular pattern (A). The immunohistochemical analysis showed positive staining for chromogranin A (B) and cytokeratin (C). The Ki67 labeling index was approximately 5% (D). All magnifications ×200.
Discussion
VHL disease is an autosomal dominant tumor predisposition syndrome caused by pathogenic variants in the VHL tumor suppressor gene on chromosome 3, with an estimated prevalence of 1 in 36,000 and a median age of onset of 26 years. 5 The clinical diagnosis of VHL syndrome is established in a simplex case (no family history) by the presence of two or more characteristic tumors, such as RH or CNS-H, ccRCC, or PGL/PCC. In an individual with a positive family history, a single such manifestation is sufficient for diagnosis. 6 Genetic testing is the diagnostic gold standard.
VHL syndrome is classified into three main types based on the specific underlying VHL gene mutation, which predicts the spectrum and risks of associated tumors (Table 1). 1 Based on the clinicopathological features and the specific genetic mutation identified in our case, it is classified as VHL type 2B, conferring a high risk for the development of PGL/PCC and ccRCC.
Characteristics of the different VHL disease subtypes.

These mutations result in the complete absence of VHL protein and are usually associated with a severe phenotype, including early onset of tumors and a higher risk of multiple organ involvement. 1
VHL: Von Hippel-Lindau; PCC: pheochromocytomas; PGL: paraganglioma; ccRCC: clear cell renal cell carcinoma.
PGL/PCCs are relatively common in clinical practice. However, up to 40% of these cases are hereditary. Our patient was a young female with a family history of intracranial tumor in her father and hypertension/adrenal disease in her sister. Based on these clinicopathological features, we strongly suspected an underlying germline mutation. Currently, more than 12 susceptibility genes have been linked to hereditary PGL/PCC, among which pathogenic variants in VHL account for approximately 4%–7% of cases. 7 The VHL gene plays a critical role in regulating cellular processes, including angiogenesis, apoptosis, and cell cycle control. Loss-of-function mutations in the VHL gene promote tumorigenesis primarily by stabilizing hypoxia-inducible factors (HIFs), which leads to the activation of pro-angiogenic and growth-related genes. Additional mechanisms independent of HIF signaling include impaired fibronectin matrix assembly, dysregulated p53 signaling, and altered microtubule stability. 1 Patients with VHL syndrome have 20%–30% lifetime risk of developing PGL/PCC. 7 CT, MRI, and positron emission tomography (PET)/CT serve as cornerstone imaging modalities. CT is often the first-line anatomical imaging technique for initial diagnosis. Notably, any adrenal lesion exhibiting post-contrast enhancement greater than 130 Hounsfield Units (HU) is highly suggestive of a PCC. 8 The CT findings in our case demonstrated these characteristic features. In recent years, functional imaging represented by PET has become a key multimodality molecular imaging technique in the assessment of PGL/PCC, which is vital for preoperative tumor localization, follow-up, and therapeutic planning. 8 Unfortunately, in the present case, this examination was not conducted prior to surgery due to economic reasons. Of note, there is a genotype-biochemical phenotype correlation in PGL/PCC. All patients of VHL exhibit elevated plasma normetanephrine levels while maintaining normal metanephrine levels, 9 a pattern that was recapitulated in our case. Interestingly, 30% of these patients show no hypertension or related symptoms. 7 This might be due to a reduced expression of tyrosine hydroxylase and phenylethanolamine N-methyltransferase (enzyme that converts norepinephrine to epinephrine) in VHL-related PGL/PCC. 9 In cases of bilateral PCCs or surgically challenging scenarios, maximal preservation of adrenal function is prioritized. In the present case, subtotal adrenalectomy was successfully performed to preserve a portion of the right adrenal gland. Long-term follow-up is essential to monitor for tumor recurrence or metastasis. Annual follow-up should include plasma metanephrines or urinary catecholamines. In addition, recent studies indicate that plasma 3-methoxytyramine, a predictor of metastatic risk, should be monitored. 10 Abdominal MRI or CT is indicated every 1–2 years. If catecholamines are elevated, PET/CT should be performed. 1 As of this writing, the patient has been free from any signs of recurrence or metastasis.
CcRCCs develop in 40%–70% of individuals with VHL syndrome, typically emerging during the third or fourth decade of life. 11 Our patient was classified as VHL type 2B, which confers a high risk for ccRCC. Despite the absence of renal lesions in the 5 years post-operation, ongoing annual surveillance with CT or MRI remains mandatory. 1 For any detected lesion under 3 cm, repeat imaging is recommended every 3 to 6 months until stability is confirmed over three consecutive scans. Conversely, the detection of any lesion measuring >3 cm warrants an immediate referral to urology. 5
A notable 8%–17% of VHL patients develop PNETs, showing a potential female predilection. 12 VHL-associated PNETs are clinically distinct from sporadic counterparts, being characterized by their typical low-grade, nonfunctional nature, the absence of symptomatic hormone hypersecretion, indolent growth, and localized presentation. 10 According to management guidelines for VHL syndrome, low-risk tumors measuring less than 1.5 cm in diameter are typically managed with active surveillance. The current case exhibited histologically low-grade features, the solitary pancreatic head tumor exceeded 3 cm in its maximum diameter, thus fulfilling the established criteria for surgical intervention.4,12
CNS-H constitute the most prevalent tumor type in VHL disease, developing in 60%–80% of patients. 5 They serve as the initial symptomatic manifestation in about 40% of cases, offering a critical diagnostic clue. Despite being histologically benign, CNS-H account for approximately 50% of deaths in VHL patients. 5 In the current case, the cerebellar nodule has remained stable and asymptomatic for 5 years postoperatively. Based on this indolent course, we recommend surveillance MRI at biennial intervals. Should radiological progression be detected, more frequent MRI examinations would be indicated, with management to be guided by neurology/neurosurgery specialists.
In addition, the clinical management of VHL syndrome is complex and typically requires a multidisciplinary approach tailored to the size, location, and symptoms of the tumors. Treatment modalities include surgical resection and targeted therapies used to control tumor growth and manage symptoms. Currently, the most promising therapeutic modalities for VHL-related disease are HIF-2α inhibitors, particularly Belzutifan. Preliminary evidence has demonstrated their efficacy in treating ccRCC, CNS-H, and PNET.12,13
Conclusion
VHL syndrome, a rare disorder, frequently has a delayed diagnosis owing to its highly variable presentation. By detailing a case of multiple neuroendocrine tumors to review VHL syndrome, this article improves diagnostic vigilance for this rare entity, even without encompassing its full spectrum.
Footnotes
Ethical approval
This study was approved by the ethics committee of Guiqian International Hospital on 30 September 2025. Approval number: GQ20250049.
Consent to participate
For this study, photos, and writing of our manuscript, the patient has given their written informed consent.
Author contributions
YC drafted the original manuscript and designed the case report. DG critically reviewed and revised the manuscript. NZ performed the literature review. YS analyzed and interpreted the radiological imaging. XH and HT made the pathological diagnosis. JX provided the clinical data. All authors have read and approved the final manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
All data generated or analyzed during the current study are included in this published article (High throughput sequencing data was performed by Shanghai Weihansi Biomedical Technology Co., Ltd).