
Abstract
Objective
The objective of this study was to compare CFTR potentiators icenticaftor versus ivacaftor (IVA) to elucidate efficacy in augmenting CFTR function while preserving CFTR protein levels.
Methods
Single-channel measurements of CFTR in the presence and absence of IVA or icenticaftor were performed with membrane vesicles from BHK-21 cells stably expressing CFTR. CFTR-expressing BHK-21 cells and non-cystic fibrosis (CF) and CF primary human bronchial epithelial cultures were treated for 48 hours with elexacaftor/tezacaftor (ELX/TEZ) plus IVA or icenticaftor and then western blot analyses were performed to assess CFTR protein maturation. Non-CF and CF primary human bronchial epithelial cultures treated with CFTR modulators were subjected to Ussing chamber analysis to evaluate CFTR functional rescue upon 48-hour treatment and acute potentiator exposure.
Results
Corrector-rescued F508del CFTR displayed increased function with icenticaftor compared to IVA by single-channel measurements. In primary F508del cultures, 48-hour treatment with ELX/TEZ plus icenticaftor led to increased CFTR function in Ussing chambers compared to treatment with ELX/TEZ plus IVA. Western blot analysis demonstrated that F508del was destabilized by IVA but not icenticaftor. Primary N1303K CFTR cultures did not exhibit enhanced rescue with icenticaftor when compared to IVA, indicating that different CFTR mutations respond differently to potentiators.
Conclusion
Icenticaftor is superior to IVA as a potentiator for ELX/TEZ-rescued F508del CFTR, as 48-hour treatment with icenticaftor enhanced F508del function but did not destabilize F508del. Understanding the mechanisms underlying CFTR potentiator activities may offer further benefits for people with CF who have F508del or other CFTR mutations.
Introduction
Cystic fibrosis (CF) is caused by mutations in the CFTR gene, which encodes for the CF transmembrane conductance regulator (CFTR) protein, a highly regulated ion channel that mediates chloride and bicarbonate transport across epithelial cells. CFTR activity is specifically important for maintaining proper mucosal surface hydration to diminish mucus concentration and allow for proper mucociliary clearance. 1 The mucosal surfaces of people with CF (pwCF) are dehydrated, which increases mucus concentration and viscosity, leading to airway plugging and decreased mucociliary clearance, resulting in a cycle of increased inflammation and susceptibility to infection.2–4 Since the identification of the CFTR gene in 1989, 5 great efforts have been made to alleviate symptoms in pwCF, largely through managing disease manifestations such as inflammation and infection. Recently, with the advent of highly effective modulator therapies (HEMT) that were developed with the help of primary CF airway cultures, substantial progress has been made in improving the health of pwCF. 6 PwCF treated with HEMTs experience notable clinical improvements in lung function as measured by forced expiratory volume in one second (FEV1), reduced inflammatory markers, decline in pulmonary exacerbations, and reported improved quality of life.6–9 CFTR modulators are components of HEMT and categorized as correctors, which promote the processing and transfer of misfolded mutant CFTR to the apical membrane of epithelial cells, and potentiators, which increase CFTR channel activity at the apical surface. The first HEMT approved by the FDA, Kalydeco,10,11 contains potentiator ivacaftor (IVA) and resulted in unprecedented improvements in the health of pwCF with CFTR gating mutations, such as G551D.12,13
Combination therapies Orkambi (IVA plus corrector lumacaftor) and Symdeko/Symkevi (IVA plus corrector tezacaftor (TEZ)) are beneficial for pwCF homozygous for the most prevalent CFTR mutation, F508del, which causes defects in CFTR protein folding and trafficking to the cell surface.14–16 Notably, Orkambi and Symdeko/Symkevi are not as beneficial for the pwCF harboring the F508del mutation as Kalydeco is for pwCF harboring gating mutations. Interestingly, in vitro research using primary human bronchial epithelial (HBE) cultures has indicated that this is due to IVA-induced increases of internalization and degradation of mature F508del CFTR.17,18 Thus, the triple combination HEMT, Trikafta/Kaftrio, was developed,7,19,20 which is composed of correctors elexacaftor (ELX) and TEZ, and potentiator IVA and is highly beneficial for pwCF with F508del and other CFTR mutations, which accounts for ∼90% of pwCF.7–9 Benefits of ELX/TEZ/IVA include sustained enhancements in lung function and nutritional status, along with a marked decrease in daily care burdens, leading to an overall improved quality of life. Indeed, recent data from in vitro and in vivo studies indicate that this triple combination surpasses the HEMT benchmark set by the IVA treatment of pwCF with G551D and other gating mutations. 6 In pwCF with one copy of F508del CFTR, ELX/TEZ/IVA improved FEV1 by 14.3%, reduced the pulmonary exacerbation rate by 63%, and improved respiratory symptoms. In pwCF with two copies of F508del, ELX/TEZ/IVA increased FEV1 by 10% above TEZ/IVA and improved respiratory symptoms.7,8
To identify improved therapies for pwCF and to understand clinical relevance of therapeutic delivery and efficacy, it is imperative to test therapeutic candidates in relevant in vitro models such as fully differentiated primary HBE cultures. After the approval of Trikafta/Kaftrio, efforts to better understand the impact of ELX/TEZ/IVA on CF pathophysiology have revealed that ELX/TEZ/IVA treatment reverses many of the aberrant properties of mucus in CF HBE, and that this impact reflects improved airway hydration. 21 Furthermore, there is now improved understanding of the pharmacological properties of these drugs. 22 However, despite these positive outcomes, infection and inflammation remain a problem23–26 and there have been reports of adverse off-target effects due to HEMT.27–29 Thus, there is still a need for development of improved CFTR-targeting therapeutics for many pwCF. This includes those who do not find sufficient relief by currently available therapies or are not eligible for HEMT because they produce insufficient CFTR protein for correction, or have CFTR mutations such as premature stop codons, splice mutations, or insertion/deletion mutations. As ELX/TEZ/IVA was found to augment the effects of splice-switching oligonucleotides 30 or readthrough reagents, 31 efforts are indeed in progress for developing new therapies, particularly for pwCF who are ineligible for HEMT, and for those who still experience disease symptoms despite CFTR modulator use.
Based on experiments in HBE cultures, treatment with ELX/TEZ/IVA led to F508del CFTR protein maturation and significant improvement in chloride transport. 32 However, there is evidence that the IVA component in this triple therapy reduces F508del rescue17,18,33 indicating that a different potentiator that does not decrease the amount of corrector-rescued F508del would be highly beneficial for pwCF to advance CFTR rescue and function. It is unlikely that the problem with F508del rescue would be resolved by utilizing deutivacaftor, a deuterated form of IVA with slower turnover, in the novel once-a-day triple treatment, Alyftrek (vanzacaftor/TEZ/deutivacaftor), which was recently approved in the United States. 34 Alleviating the destabilization of rescued F508del may be achieved with novel CFTR potentiator compounds that act on rescued F508del without decreasing F508del protein levels and channel function at the apical membrane. In this study we tested the effects of the novel CFTR potentiator compound icenticaftor/QBW251 (ICN), which is currently in clinical trials for chronic obstructive pulmonary disease (COPD),35–38 for its ability to enhance rescued CFTR function.
Materials and methods
Epithelial culture
Primary HBE cells containing predominantly large airway cells from proximal sites of adult male and female donors were obtained from the UNC Marsico Lung Institute (MLI) Tissue Procurement and Cell Culture Core and then seeded on 12 mm Snapwell inserts (Corning; Corning, NY) coated with human placenta collagen (Sigma; St Louis, MO) at 250,000 cells/insert for 5 to 7 days until air-liquid interface was achieved and then cultures were maintained using a modified Lonza differentiation medium on the basolateral side that includes a 1 : 1 ratio of BEBM (Lonza; Walkersville, MD) and DMEM (Thermo Fisher Scientific; Waltham, MA) plus BEGM SingleQuots (Lonza) as described elsewhere 39 for 3 to 4 weeks to allow cells to become fully differentiated as previously described.40,41 Donor information: (1) 52-year-old Caucasian male non-CF nonsmoker, (2) 45-year-old Caucasian male non-CF nonsmoker, (3) 37-year-old Black male non-CF nonsmoker, (4) 42-year-old Black female non-CF nonsmoker, (5) 30-year-old Caucasian female with CF (F508del/F508del), (6) 51-year-old Caucasian male with CF (F508del/F508del), (7) 30-year-old Caucasian male with CF (F508del/F508del), (8) 32-year-old Caucasian female with CF (F508del/F508del), (9) 42-year-old Caucasian male with CF (N1303K/394delTT).
BHK-21 clone 13 (RRID:CVCL_1915; ATCC catalog number: CCL-10) derived from baby hamster kidney cells were grown in DMEM/F12 media with 5% fetal bovine serum and 500 μM methotrexate to select for stably expressed CFTR as described previously. 42
Drug treatments
A CFTR corrector mixture containing two correctors, ELX (3 μM) plus TEZ (10 μM), was applied alone or with potentiator IVA (5 μM) or ICN (5 μM) (Selleck Chemicals; Houston, TX) to primary HBE cultures on the apical and basolateral sides for 48 hours. For BHK-21 dose response experiments, ELX (3 μM) and TEZ (10 μM) were added to the media, with IVA or ICN added at 0.1, 1, 2, or 5 μM for 48 hours. For BHK-21 cells used for single-channel experiments, each CFTR modulator was added to cell culture media at 2 μM for 24 hours. For vehicle controls, dimethyl sulfoxide (DMSO), was added to the cell culture media at volumes identical to CFTR modulator volumes.
Single-channel measurements
Membrane vesicles from BHK-21 cells stably expressing wild-type (WT) or F508del CFTR were isolated and single-channel recordings were obtained as previously described. 43
Western blot analysis
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting of BHK-21 cells stably expressing WT or F508del CFTR were performed as previously described. 43 Briefly, total cell lysates were separated on 4% to 20% gradient gels and then proteins were transferred to nitrocellulose (Bio-Rad; Hercules, CA) and probed with CFTR-specific monoclonal antibody 596 (1 : 1000; CFTR Antibody Distribution Program; https://cftrantibodies.web.unc.edu). Primary HBE cells were lysed and then CFTR proteins were immunoprecipitated and then western blot analysis was performed using primary monoclonal antibodies specific for CFTR protein as previously described. 17 Briefly, whole-cell lysates were prepared and then CFTR was immunoprecipitated using rabbit anti-CFTR polyclonal antibody 155 (1 : 200; generously provided by Dr John R. Riordan). Samples were separated on 4% to 20% gradient SDS-PAGE gels (Bio-Rad) and then transferred to nitrocellulose. Blots were probed with mouse monoclonal anti-CFTR antibodies 596 and 217 (1 : 2000 each; CFTR Antibody Distribution Program) and then with Alexa Fluor 680-conjugated goat anti-mouse secondary antibody (1 : 10,000; Thermo Fisher, catalog number A-21058). Rabbit anti-actin antibodies (1 : 4000; Cell Signaling; Beverly, MA, catalog number 4970) were used as a loading control and detected using Alexa Fluor 790-conjugated goat anti-rabbit secondary antibody (1 : 20,000; Thermo Fisher, catalog number A11369). Protein bands were visualized using a Sapphire Biomolecular Imager (Azure Biosystems; Dublin, CA) and quantitated with AzureSpot software (version 2.2.170, Azure Biosystems).
Ussing chamber measurements
HBE cultures were mounted in Ussing chambers (Physiological Instruments) to measure ISC as previously described. 17 HBE cultures in Ussing chambers were equilibrated to 37° C in a bilateral bath of Krebs-bicarbonate-Ringer buffer (KBR; in mM: 140 Na + , 120 Cl−, 5.2 K + , 1.2 Ca2 + , 1.2 Mg2 + , 2.4 HPO42−, 0.4 H2PO4−, 25 HCO3−, and 5 glucose, pH 7.4) and circulated with 95% O2 to 5% CO2. Short-circuit currents (ISC) were measured as previously described.44–46 Amiloride (100 µM) was added to block the epithelial sodium channel, ENaC, followed by forskolin (10 µM) to stimulate CFTR. CFTR potentiators IVA or ICN were added acutely to cultures in Ussing chambers as a single dose at 1 µM after forskolin, or as a dose response before or after forskolin at 0.1, 1, 2, and 5 µM. CFTR was then inhibited with CFTRinh-172 (10 µM). Data were analyzed using Acquire and Analysis (version 2.3) software (Physiologic Instruments). ISC traces were imported to and processed in Origin 2024 (10.1) (OriginLab Corporation).
Statistics
Results are presented as means of average responses per BHK-21 replicate or HBE donor, and error bars show SEM. Normal distributions were checked by Shapiro-Wilk normality test. If the values passed, the two-tailed Student's t-test was used, and if not, the Mann-Whitney test was used. Statistical analyses were performed in GraphPad Prism (version 10.4.0, GraphPad Software; Boston, MA). The p-values of < 0.05 indicate statistical significance. This study was conducted in accordance with the Helsinki Declaration of 1975 as revised in 2024.
Results
Icenticaftor acts as a potentiator for WT CFTR function by single-channel measurements and Ussing chambers
Icenticaftor/QBW251 (ICN) (Figure 1(a), left 35 ) is a CFTR potentiator that is currently being tested in clinical trials for COPD.35–38 IVA (Figure 1(a), right 47 ), is a well-known CFTR potentiator that is an active component of the HEMTs for pwCF, Kalydeko and Trikafta/Kaftrio. Although IVA was proven to function extremely well as a CFTR potentiator, it has also been shown to destabilize corrector-rescued F508del.17,18,33 To examine the potentiator activity of ICN compared to IVA, we first looked at CFTR function in single-channel measurements from BHK-21 cells stably expressing WT CFTR. WT CFTR open probability (Po) was similarly improved after addition of ICN or IVA (Figure 1(b)). Although the Po increased upon the addition of potentiators, the channel conductance (γ; pS = picosiemens) remained very similar, which implies that although the channels are spending different amounts of time in the open state, each channel can conduct chloride ions when open.
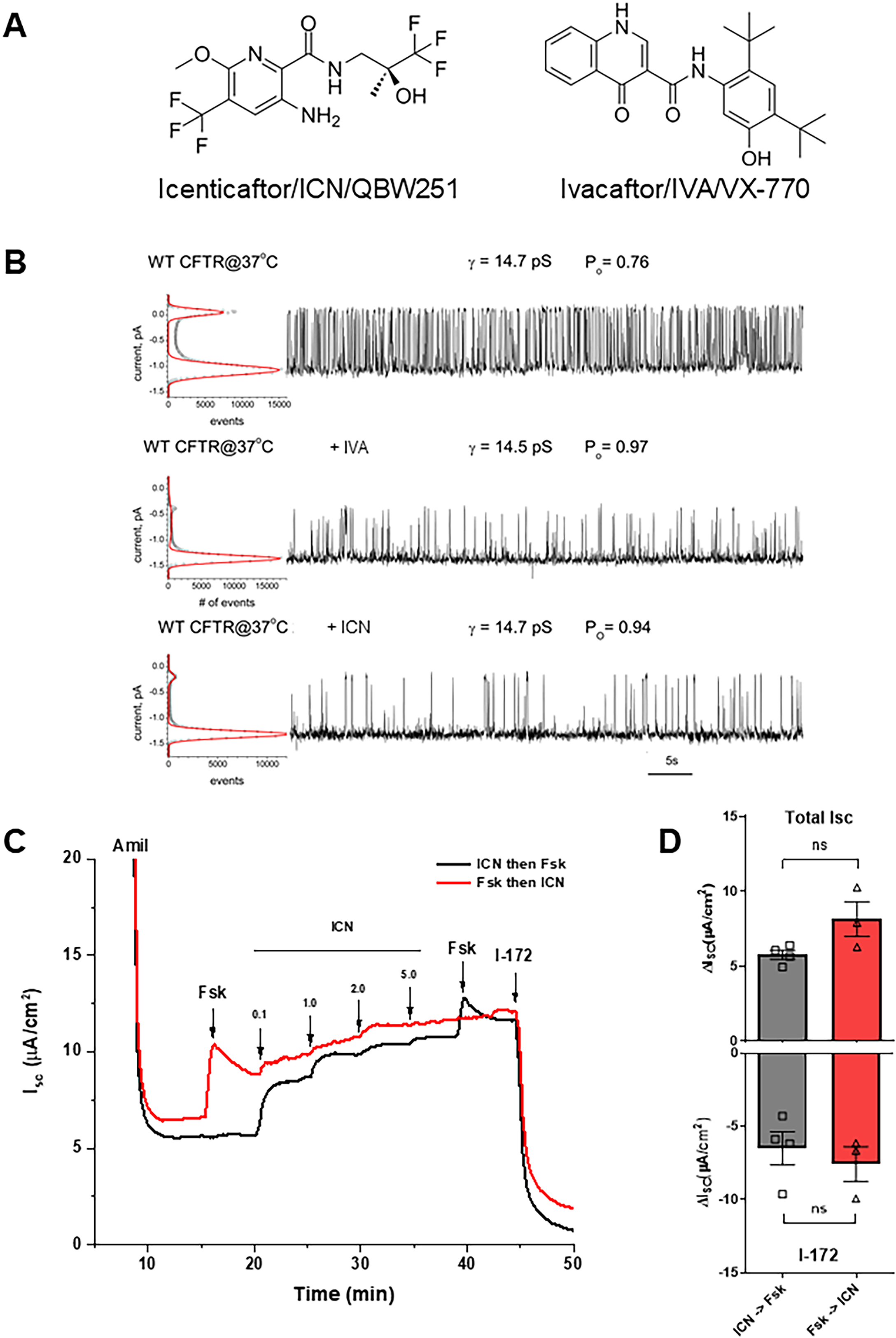
Icenticaftor (ICN) acts as a potentiator for wild-type (WT) CFTR function by single-channel measurements and Ussing chambers. (a) Chemical structure of potentiators icenticaftor (ICN, QBW251; left) and ivacaftor (IVA, VX-770; right). (b) Single-channel measurements of WT CFTR untreated (top), treated with IVA (2 μM, middle), or ICN (2 μM, bottom). (c) Representative short-circuit current (ISC) traces from measurements in Ussing chambers depicting a dose response for ICN in non-cystic fibrosis (CF) HBE where either ICN is administered before forskolin (Fsk; black) or Fsk is administered before ICN (red). (d) Quantitation of responses shown in (c). Top: total sum of short-circuit currents (ISC) upon addition of Fsk plus ICN (Total ISC). Bottom: change in ISC upon addition of CFTR inhibitor, I-172. Statistical analyses were performed by unpaired two-tailed Student’s t-test; ns = not significant, mean ± SEM, n = 3–4 donors, 2–4 cultures per donor.
We next examined the effects of ICN on WT CFTR function in primary HBE cultures. Non-CF primary HBE cultures from four donors were mounted in Ussing chambers and then treated with increasing doses of ICN. ICN enhanced the function of WT CFTR in a dose-dependent manner, starting at concentrations as low as 0.1 μM, before (black trace) and after (red trace) addition of forskolin (Fsk) (Figure 1(c)). Short-circuit currents (ISC) showing total stimulation with ICN and Fsk (Total ISC) and upon addition of CFTR-specific inhibitor, I-172, were quantitated from four non-CF HBE donors (Figure 1(d)). There was no difference between CFTR function whether Fsk was added before or after ICN, suggesting that potentiation by ICN does not require WT CFTR to be phosphorylated.
ICN is a superior potentiator over IVA for F508del and does not destabilize F508del
To examine the effects of ICN compared to IVA on F508del CFTR function and stability, we first performed single-channel measurements with membranes of BHK-21 cells stably expressing F508del CFTR. F508del CFTR treated with correctors but without potentiator displayed negligible channel activity (Po = 0.05; Figure 2(a), top). ICN (Figure 2(a), bottom) strongly increased the Po of F508del to more than double the Po of IVA (Figure 2(a), middle) (Po = 0.74 vs. 0.32). As was seen with WT CFTR single-channel measurements (Figure 1(b)), although there were differences in F508del CFTR Po with different treatments, the channel conductance (γ) remained very similar. Western blot analyses using CFTR-specific antibodies demonstrated CFTR protein levels and maturation from whole-cell lysates (Figure 2(b) and (d)). WT CFTR (Figure 2(b); WT) stably expressed in BHK-21 cells processed high levels of the mature form of CFTR, band C (★). In BHK-21 cells stably expressing F508del treated with vehicle (Figure 2(b); vehicle) only the immature form of CFTR, band B (•), was detected. In BHK-21 cells expressing F508del treated with ELX/TEZ for 48 hours, the mature form was detected. Upon 48-hour treatment of ELX/TEZ and increasing doses of IVA or ICN, as shown, there was a significant decrease in the ratio of band C to band B (C/B ratio) upon treatment with IVA at 5 μM that did not occur with ICN at 5 μM (Figure 2(b) and (c)). In fully differentiated primary CF HBE cultures homozygous for F508del derived from four donors (Figure 2(d)) pretreated with CFTR modulators, IVA caused a significant destabilization of mature F508del (★) that did not occur with ICN (Figure 2(e)). This decrease in mature CFTR band C levels in HBE lysates (Figure 2(d):★ and (e)) suggests that less processed CFTR is available with IVA compared to ICN, highlighting a possible impact on CFTR function in airway epithelia.

Icenticaftor (ICN) is a superior potentiator over ivacaftor (IVA) for F508del and does not destabilize F508del. (a) Single-channel recordings of drug-rescued (dr) CFTR incorporated into membrane vesicles prepared from BHK-21 cell lines stably expressing F508del CFTR in the preformed lipid bilayer. Top: treatment with corrector elexacaftor (ELX, 2 μM) combined with tezacaftor (TEZ, 2 μM). Middle: treatment with ELX (2 μM) plus IVA (2 μM). Bottom: treatment with ELX (2 μM) plus ICN (2 μM). Treatment with ICN results in a channel open probability (Po) of 0.74 that is more than twice as high as treatment with IVA (0.32). (b) and (d) Western blots of whole-cell lysates using CFTR-specific antibodies to detect CFTR levels and maturation; mature CFTR band C (★) and immature band B (●). Actin is shown as a loading control. (b) BHK-21 cells expressing WT (untreated) or F508del CFTR treated with vehicle or correctors ELX (3 μM) and TEZ (10 μM) plus different doses of potentiators ICN or IVA, as shown, for 48 hours. (c) Quantitation of the ratio intensities of band C to band B (C/B ratio) from panel B. Statistical analyses were performed by unpaired two-tailed Student’s t-test; *p < 0.05, ns = not significant, mean ± SEM, n = 3 replicates. (d) Representative blot from primary human bronchial epithelial (HBE) cultures from a cystic fibrosis (CF) donor (F508del/F508del) grown at air-liquid interface and treated with CFTR modulators: ELX (3 μM) and TEZ (10 μM), + /− IVA or ICN (5 μM each) for 48 hours. (e) Quantitation of the C/B ratio from panel D. Statistical analyses were performed by unpaired two-tailed Student’s t-test; *p < 0.05, ****p < 0.0001, ns = not significant, mean ± SEM, n = 4 donors, 4 cultures per donor.
We next examined the effects of different doses of ICN or IVA on corrector-rescued F508del CFTR function in primary HBE cells. Primary CF HBE cultures from three donors homozygous for F508del pretreated with ELX/TEZ for 48 hours were mounted in Ussing chambers and then exposed to increasing doses of ICN or IVA after (Figure 3(a) and (b)) or before (Figure 3(c) and (d)) Fsk. ICN and IVA enhanced F508del CFTR function in a dose-dependent manner, starting at concentrations as low as 0.1 μM, as was seen with WT CFTR (Figure 1(c)). ISC showing total maximum stimulation with ICN or IVA and Fsk (Total Max Stim) and upon addition of I-172, were quantitated from three CF (F508del/F508del) HBE donors (Figure 3(b) and (d)). There was an increase (although not significant) in F508del CFTR function with ICN versus IVA when potentiators were added before Fsk (Figure 3(c) and (d)) and a significant increase in response to I-172 with ICN versus IVA (Figure 3(c) and (d)), indicating that potentiation by ICN does not require F508del CFTR to be phosphorylated, unlike IVA. 47

Primary cystic fibrosis (CF) (F508del/F508del) human bronchial epithelial (HBE) cultures treated with icenticaftor (ICN) or ivacaftor (IVA) before or after forskolin in Ussing chambers reveal that ICN activity does not require CFTR to be phosphorylated. (a) Representative ISC traces from measurements in Ussing chambers depicting a dose response for ICN or IVA in CF (F508del/F508del) HBE cultures where Fsk was administered before potentiators. HBE cultures were pretreated continuously (c) for 48 hours with correctors elexacaftor (ELX; 3 μM) and tezacaftor (TEZ; 10 μM) and then increasing concentrations of potentiators ICN or IVA were administered acutely (a) in Ussing chambers. (b) Quantitation of responses shown in (b). Top: total maximum stimulation (Total Max Stim) of ISC upon addition of Fsk and ICN or IVA. Bottom: change in ISC upon addition of I-172. Statistical analyses were performed by unpaired two-tailed Student’s t-test; ns = not significant, mean ± SEM, n = 3 donors, 4 cultures per donor. (c) Representative ISC traces from measurements in Ussing chambers depicting a dose response for ICN or IVA in CF (F508del/F508del) HBE cultures where potentiators were administered before Fsk. HBE cultures were pretreated with correctors as described for panel A. (d) Quantitation of responses in panel C, as described for panel B; *p < 0.05, ns = not significant.
Ussing chamber data using primary CF (F508del/F508del) HBE cells treated for 48 hours with ELX/TEZ/ICN led to a superior functional rescue when compared to ELX/TEZ/IVA (Figure 4). This result is consistent with the results of F508del CFTR single-channel experiments and western blot analyses after 48-hour treatment with ELX/TEZ plus ICN vs IVA (Figure 2). Figure 4(a) shows representative Ussing chamber traces. Treatment with ELX/TEZ plus IVA for 48 hours resulted in a significant decrease in Fsk response compared to correctors alone (ELX/TEZ), while ELX/TEZ plus ICN did not result in a significant decrease compared to ELX/TEZ alone (Figure 4(b)). Note that this pattern matches F508del HBE western blot data (Figure 2(e)). The addition of acute (a) IVA or ICN treatment combined with the Fsk response is displayed as the total maximum stimulation (Total Max Stim; Figure 4(c)). Acute treatment with ICN was similar to IVA after 48-hour treatment with ELX/TEZ. CFTR specificity is confirmed with CFTR inhibitor, I-172 (Figure 4(d)), showing a reflective pattern of Figure 4(c). Together, these data indicate that 48-hour treatment with ICN has a stronger effect on corrector-rescued F508del CFTR function than IVA and unlike IVA, ICN does not destabilize mature F508del CFTR. ICN is therefore superior over IVA as a potentiator for corrector-rescued F508del CFTR.

Rescued F508del CFTR showed an increase in function with ICN compared to IVA in primary human bronchial epithelial (HBE) cultures. (a) Ussing chamber measurements of primary cystic fibrosis (CF) (F508del/F508del) HBE cultures after treatment with CFTR modulators continuously (c) for 48 hours as described in Figure 2(d). Quantitation of ISC upon administration of (b) Fsk peak, (c) Total Max Stim between Fsk and acute (a) IVA or acute (a) ICN (1 μM each), and (d) I-172 in Ussing chambers. Statistical analyses were performed by unpaired two-tailed Student’s t-test; *p < 0.05, **p < 0.01, ***p < 0.001, ns = not significant, mean ± SEM, n = 4 donors, 4 cultures per donor.
Treatment with ICN may not be as beneficial for CFTR rare mutation, N1303K
We next tested whether the effects of ICN were also seen in a different CFTR mutation. N1303K CFTR is a folding/processing mutation like F508del but has defects in other folding pathways. 48 Primary CF HBE cultures harboring the genotype N1303K/394delTT were treated with various combinations of CFTR modulators (Figure 5), as was done in Figure 4. Representative Ussing chamber traces are shown in Figure 5(a). Treatment for 48 hours with corrector compounds ELX and TEZ, combined with the potentiator ICN or IVA led to a significant increase N1303K CFTR rescue with IVA compared to ICN (Figure 5(b)). When treated acutely with ICN or IVA, IVA was more beneficial for N1303K than ICN (Figure 5(c) and (d)). These results for N1303K are different from what was observed for F508del, suggesting that potentiators act in a CFTR mutation-specific manner.

Treatment with ICN may not be as beneficial for CFTR rare mutation, N1303K. CF (N1303K/394delTT) human bronchial epithelial (HBE) cultures were treated continuously (c) for 48 hours with CFTR modulators as described in Figure 2(d) and then ISC were measured in Ussing chambers with acute (a) administration of CFTR modulators as described in Figure 4 to examine CFTR function. (a) Representative Ussing chamber traces in response to various agonists and antagonists. Quantitation of ISC in response to (b) Fsk, (c) Total Max Stim, (d) I-172. Statistical analyses were performed by a Mann-Whitney test; *p < 0.05, ns = not significant, mean ± SEM, 1 donor, n = 4 replicates.
Discussion
CF shares several pathophysiological pulmonary features with COPD, including mucus obstruction of the small airways, mucus hypersecretion, airway inflammation, and chronic bacterial infection.49–51 As smokers have an increased risk of CFTR dysfunction, and impaired CFTR function leads to worsening of COPD symptoms, CFTR is being explored as a novel target and the use of CFTR modulators in COPD is now under active investigation.52–54 It was suggested that insights into the mechanisms of CFTR function may guide the development of advanced treatments for specific patient subgroups. 54
ICN is currently being tested in clinical trials for people with COPD to reduce excess mucus and was found to significantly improve FEV1 over placebo in patients with moderate-to-severe COPD and chronic bronchitis. 38 ICN demonstrated a statistically significant reduction in systemic inflammation and showed a reduction in bacterial colonization in exploratory sputum colony forming unit analysis. ICN was well tolerated with a similar incidence of adverse events between ICN and placebo groups, and was suggested to qualify as candidate novel therapeutic approach in COPD that may provide clinically relevant benefits warranting further investigation in future clinical trials. 38 The safety and efficacy of ICN was also tested in pwCF with CFTR mutations that affect protein folding or gating/channel function. 36 ICN was found to be safe and well-tolerated in healthy volunteers and pwCF, with no unexpected events or discontinuations in CF groups, while showing clinically meaningful improvements in lung function (mean %predicted FEV1 increased by 6.46%), LCI 2.5 (decreased by 1.13 points), and sweat chloride (decreased by 8.36 mmol/L) in pwCF with CFTR gating mutations, but no significant improvements in pwCF homozygous for F508del CFTR. These results suggest that, as was previously observed with IVA, ICN monotherapy is not sufficient for pwCF with folding mutations such as F508del, highlighting the need for combination therapy with at least one CFTR corrector to achieve clinical benefit for individuals harboring CFTR folding mutations. Given its demonstrated success in pwCF with CFTR gating mutations, ICN appears promising as part of a combination therapeutic approach with CFTR correctors for these patients. 36 This is in accordance with the observation that ICN-enhanced activity of the CFTR gating mutant, G551D. 35 This group also suggested treatment with ICN for smoking-induced dysregulation of CFTR 35 following the observation that cigarette smoke decreased CFTR protein and function. 55 A recent phase 2 clinical trial using ICN demonstrated benefits in lung function, symptoms, quality of life outcomes, and fibrinogen concentrations for people with COPD who already received maximal inhaled therapy. 37 Furthermore, as ICN was well tolerated and the results were consistent, a reasonable dose of ICN (300 mg twice daily) to manage symptoms and exacerbations in people with COPD was identified. 37 In a study that examined the absorption, distribution, metabolism, and excretion of a single dose of radioactive ICN, ICN was found to be sufficiently metabolized and demonstrated good systemic availability, thereby supporting further development of ICN as a therapeutic. 56 ICN was tested for electrocardiogram parameters and was found to have no clinically relevant effects. 57
The doses of ICN used in the current study are relevant as they correspond to approximately half of the peak plasma concentrations observed with chronic administration, and about five times lower than peak levels seen with acute dosing. 56 As previously discussed,17,58,59 the concentrations of ICN employed here also fall within the range of plasma concentrations reported in clinical studies.15,32 Although ICN was suggested as a candidate for further therapeutic development for patients with COPD, 37 the results of our current study suggest that ICN is a much better potentiator for F508del than for WT CFTR when compared to IVA (Figures 1–4).
WT CFTR open probability (Po), which is the fraction of time the channel remains open, was similarly increased by ICN and IVA in single-channel recordings (Figure 1(b)). However, the conductance was not affected upon potentiator treatment with ICN or IVA (Figure 1(b)). This indicates that ICN, similar to IVA may support the same open channel conformation that allows anions to flow through the pore. 60 Interestingly, IVA was suggested to require CFTR to be phosphorylated 47 in order to potentiate CFTR; however, our data suggest that ICN enhances CFTR function independently of phosphorylation, as its effect is evident even without forskolin stimulation (Figures 1(c) and (d) and 3(c) and (d)). Thus, unlike IVA, ICN can potentiate WT and F508del CFTR in a phosphorylation-independent manner (Figures 1(c) and (d) and 3(c) and (d)).
We examined single-channel activity of corrector-rescued F508del CFTR and found that treatment with ICN resulted in an increased Po compared to IVA (Figure 2(a)). When combined with correctors ELX and TEZ, 48-hour ICN treatment of BHK-21 and primary HBE did not destabilize corrector-rescued F508del CFTR, unlike IVA (Figure 2(b)–(e)). A 48-hour treatment with ICN further increased F508del function of primary HBE in Ussing chambers compared to IVA (Figure 4(b)), which corresponds to levels of CFTR maturation with these same treatments (Figure 2(e)).
We tested whether these enhanced effects of ICN compared to IVA were also seen in the rare CFTR mutation, N1303K (Figure 5), which is approved for ELX/TEZ/IVA treatment, but has shown only little improvement in CFTR function when compared to F508del.61–63 Using primary HBE harboring the N1303K CFTR mutation, 48-h treatment with corrector compounds ELX and TEZ with potentiator ICN or IVA led to a significant increase in N1303K CFTR rescue with IVA (Figure 5(b)), which is the opposite of what occurred with F508del where 48-hour treatment with correctors plus IVA led to a decrease in CFTR function (Figure 4(b)). When treated acutely in Ussing chambers with ICN or IVA, IVA was more beneficial for N1303K than ICN (Figure 5(c) and (d)), which again, is different from what we found for F508del CFTR where acute treatments with ICN or IVA led to a similar increase in CFTR activity (Figure 4(c) and (d)). Although the N1303K experiment was performed from only one N1303K donor (Figure 5), which is a limitation of this study, these differences between N1303K and F508del responses to ICN or IVA suggest that potentiators act in a CFTR mutation-specific manner. Like F508del, N1303K CFTR is a folding/processing mutation. 48 However, N1303K and F508del differ in their folding, processing, intracellular handling, and gating patterns,48,64–67 which may explain these unique responses to potentiators. This personalized outcome encourages testing of efficacy of ICN in other CFTR mutations. These studies were conducted in the well-established HBE model.40,41 Recently, HBE models of inflammation and infection have been developed.68,69 As inflammation and chronic bacterial infections are present in the airways of pwCF and COPD, future studies examining the efficacy of ICN may include HBE that are inflamed and/or infected.
Conclusion
In an effort to identify a potentiator with optimized activity that lacks detrimental effects on CFTR, based on our results, we can conclude that ICN is a superior potentiator for F508del CFTR when compared to IVA as 48-hour treatment with ICN enhanced F508del function but did not destabilize corrector-rescued mature F508del, thus leading to improved function. We also examined the effects of ICN on the rare CFTR mutation, N1303K. Although F508del and N1303K are both folding/processing mutants, the benefits of 48-hour ICN treatment appear to be more pronounced for F508del than for N1303K. In contrast, acute treatment with IVA may lead to a higher level of function for N1303K than acute treatment with ICN. These results suggest that CFTR potentiator activity is mutation specific. This information is critical for considering development of advanced therapies as understanding the mechanisms of different CFTR potentiator activities may allow for optimization of therapies for individuals, thereby providing increased benefits for pwCF with F508del, N1303K, and other CFTR mutations. Future studies will address whether ELX/TEZ/ICN results in enhanced rescue of other rare CFTR mutations.
Footnotes
Acknowledgements
Ethical considerations
The primary HBE cells were obtained by the UNC MLI Tissue Procurement and Cell Culture Core. This Core uses an IRB-approved protocol (No. 03-1396) to isolate airway cells. These cells are distributed for research. All research activities complied with ethical regulations.
Consent to participate
Lung tissue obtained for this project was distributed by The Tissue Procurement and Cell Culture Core under IRB protocol #03-1396. All samples were provided as de-identified tissue, and only the age, sex, race, and CFTR genotype was provided to the user. Informed consent was not required, as the study did not involve living human subjects.
Authors’ contributions
DMC, NLQ, AAA, and MG were involved in conceptualization; DMC, LAA, NLQ, SEB, TJJ, AAA, and MG in methodology and formal analysis; DMC and MG in writing—original draft preparation; DMC, LAA, NLQ, SEB, TJJ, AAA, and MG in writing—review and editing; and MG in project administration and funding acquisition.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by CFF Research Development Program [RDP BOUCHE19R0], [ESTHER24R0]; and NIH NIDDK Cystic Fibrosis Research and Translation Center [P30DK065988].
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.