
Abstract
Bluetongue is a serious non-contagious vector-borne viral disease in ruminants, causing poor animal welfare and economic consequences globally. Concern has been raised about the development of novel bluetongue virus (BTV) strains and their possibly altered virulence through the process of viral reassortment. Virulence is traditionally estimated in lethal dose 50 (LD50) studies in murine models, but agreement with both in vitro and virulence in ruminants is questionable, and a refined experimental design is needed. Specific reassortants between wild-type and vaccine strains of BTV-1, -6 and -8 have previously been developed by reverse genetics. The aim of the present study was to rank the in vivo virulence of these parental and reassortant BTV strains by calculating LD50 in a murine model by using an experimental design that is new to virology: a between-patient optimised three-level response surface pathway design. The inoculation procedure was intracranial. Fifteen suckling mice were used to establish LD50 for each strain. Three parental and five reassortant virus strains were included. The LD50s varied from of 0.1 (95% confidence interval (CI) 0–0.20) to 3.3 (95% CI 2.96–3.72) tissue culture infectious dose 50/ml. The results support the hypothesis that reassortment in BTV may lead to increased virulence in mice with potential negative consequences for the natural ruminant host. The ranking showed low agreement with in vitro properties and virulence in ruminants according to existing literature. Refined design such as response surface pathway design was found suitable for use in virology, and it introduces significant ethical and scientific improvements.
Introduction
Bluetongue (BT) is a serious non-contagious, vector-transmitted disease in ruminants caused by BT virus (BTV), with negative animal welfare and economic consequences globally. 1 Assessment of the phenotypic properties, such as virulence, of BTV is necessary in basic studies on transmission, pathogenesis and the immune response, for production of vaccines and for development of diagnostics. In principle, viral phenotypic properties can be evaluated in vitro and in vivo. The use of live animal experiments in BT research was reviewed by Coetzee et al. 2 Large ruminants are often difficult to work with due to size, costs and possible biosafety/biosecurity issues. Murine models are therefore widely used, especially suckling mice, as they are highly susceptible to BTV via the intracranial route when younger than 2 weeks of age. Adult transgenic INFAR(–/–) mice are also susceptible to BTV infection and have been used in recent years. 3 Despite the common use of in vitro studies and murine studies for the evaluation of BTV virulence, several authors have reported a lack of correlation between findings in different models.4,5 Extrapolation of results to the natural host is also a problem. Findings even differ between ruminants due to individual differences, breed, animal husbandry and environmental factors. 4
Among the many factors that influence the in vivo experimental virulence is the choice of experimental design. Details of different design used are often poorly reported in the field of virology. A classical parallel design is traditionally chosen with 5–10 mice in each group, while the World Health Organization (WHO) guidelines for vaccine evaluation generally recommend 10 males and 10 females per group. 6 If a fixed dose is used, it is unknown if this represents the dose of interest. To ensure this, serial dilutions of the inoculum are given to groups of mice. Control groups are included, and the trial is often repeated three times. Large numbers of mice are therefore required, and many are used to test doses out of range. Mortality, usually at a fixed time point post infection, is registered, and the viral lethal dose 50 (LD50) values are calculated using the methods of Spearman–Karber 7 or Reed–Muench. 8 In toxicological LD50 estimation, this approach is no longer recommended from ethical, statistical and cost perspectives,9,10 and study designs with more accurate sequential dosing such as ‘up and down’, ‘random walk’ and ‘response surface pathway’ (RSP) are used.10,11 Basic RSP is based on the theory of stochastic chain models, where the procedure for generating the next step is based on the response at the previous one. RSP consists of several levels, with a fixed number of animals used on each level. The dose window is limited by the maximum and minimum possible or interesting dose. The first dose is often the mid-dose of the dose window. The number of animals that die at this level will direct the dose to be used at the next level through use of a standard formula. Introduction of a k-adjustment factor ensures coverage of the entire dose window.11–16 The optimised RSP design has been successfully used in toxicology, 11 and is also used in dose-finding studies in other fields.12–16
Virulence studies are necessary to estimate the potential risks associated with genetic changes of viruses. BTV has a segmented genome, and simultaneous infections with two or more virus strains can result in virus offspring with genome segments from both ‘parental’ virus strains – so-called reassortants – that may differ from their parental virus strains in phenotypic characteristics such as altered virulence. 17 Studies of BTV reassortants in cell culture 18 have supported changes in phenotype and possibly implications for the virulence and/or transmissibility of reassortant viruses in vivo. 2
In light of the possibility that poor experimental design could contribute to a lack of agreement between in vitro mice and ruminant findings in BTV studies and the overall need for refined experimental design within the field of virology, it was regarded as valuable to test an improved experimental design. The aim of the present study was to rank the in vivo virulence of parental and reassortant BTV strains using a murine model and a between-patient optimised three-level RSP design.
Methods
A live animal experiment with suckling mice as the animal model, intracranial inoculation as the experimental procedure and a between-patient three-level RSP design was chosen.11–16 Eight virus strains were evaluated: three parental and five reassortant. In total, 120 suckling mice were used (n = 15 per virus strain).
Ethics approval was obtained prior to the trial from the animal ethics committees of the University of Pretoria (AUCC; project number V059-11) and the Agricultural Research Council – Onderstepoort Veterinary Institute (ARC-OVI; project number V07911, AEC 07.13). The research was conducted under the auspices of the University of Pretoria, whereas the Transboundary Animal Diseases Facility at the ARC-OVI provided the BSL-3 facilities for the animal work.
Housing, animals and animal husbandry
The BSL-3 animal facility does not continuously house rodents. Only one experiment takes place at a time, with cleaning and disinfection between experiments. The only room that can accommodate rodents is 25 m2, with an anteroom and standard BSL-3 quality system for biosecurity such as cleaning, disinfection and environmental microbiological monitoring.
Specific pathogen free (SPF) Balb/C mice (2 days old, body weight 1–2 g) were procured as families (12 dams with 10 pups each, runts excluded) from a commercial source (Onderstepoort Biological Products, Pretoria, South Africa). Mixed sexes (male or female) were used.
The animals were transported in cardboard boxes with one family per box by motor vehicle for approximately 15 minutes to the research facility where they were transferred to cages and housed overnight, prior to labelling and inoculation. Each mouse family was housed in a wire-topped rodent breeding cage (18 cm × 30 cm × 11 cm; Lasec Laboratory Solution Provider, Johannesburg, South Africa). Absorbent aspen bedding with paper shavings to a depth of about 2 cm was provided, and rodent chew sticks (Gnaw Pucks®; Lomir Biomedical, Memphis, TN) were given as environmental enrichment. The animals were fed commercial rodent pellets (PicoLab® Mouse Diet 2058, St. Louis, MO), and potable water was supplied ad libitum. A 12-hour light/dark cycle was used to simulate a diurnal cycle, with light intensity at 323 lux during the light cycle. Atmospheric pressure was 40 Pa, humidity 55% and room temperature 21–23ºC (±1ºC). Cages were cleaned on a weekly basis.
Initially, several methods to label the mice were tested, since the randomisation required individual labelling. The final solution was toe clipping at 2 days of age. The mice received a general analgesic (Temgesic, buprenorphine, 0.05 mg/kg subcutaneously), and a topical analgesic (lidocaine 100 mg/ml containing epinephrine) was further applied to the amputation site in order to reduce pain and bleeding.
The mice were monitored for clinical signs and/or mortality in the morning (9:00am) and afternoon (3:00pm) for the duration of each trial. Clinical observations were recorded systematically by use of score sheets developed for assessment of health and welfare of mice in the relevant age span.19,20 The mice pups were inspected where possible while still in their cages in order to reduce potential stress due to handling. The humane end point was defined as the point when mice had stopped nursing, showed neurological signs (i.e. in-coordination or myoclonus) and/or wandered out of the nest. Mice were euthanised by cervical dislocation if they survived the experimental period or reached the humane end point.
Virus strains and inoculum preparation
The three parental virus strains had previously been evaluated for their in vitro properties, 18 and their virulence in ruminants was known from both experimental studies and field experience. Serotype 1, 6 and 8 (P1, P6 and P8) were used as parental strains; five virus strains were reassortants (R) of vaccine/attenuated and field/virulent strains. The R strains were named according to the nomenclature suggested by Meiring et al., 21 and their phenotype properties were known from cell culture studies. 18 All virus strains were kindly provided by Dr. P. Van Rijn and Dr. R. Van Gennip at the Central Veterinary Institute, Lelystad, the Netherlands. The strains were generated by reverse genetics as described by Boyce and Van Gennip.22,23 The background information of the parental strains and the origin of each of the 10 virus segments of each reassortant are shown in Table 1.
Overview of bluetongue virus strains used in the study: parental origin of the 10 genome segments and their encoded proteins are shown in each of the parental (P) and reassortant (R) virus strains produced by reverse genetics.

aNomenclature according to Meiring et al. 21
bDenotes reassortant strain with segments 1,3,4,8 and 9 from parental serotype 6 and other segments from parental serotype 1.
Virus stocks were prepared in Vero cells using standard culture methods 24 and concentrated from clarified cell culture supernatant using PEG 6000 precipitation (cat. # K904-50/200; non-toxic reagents; BioVision, Inc., Milpitas, CA). Viral pellets were re-suspended in 1 ml Dulbecco’s modified Eagle’s medium, and 1 M KCl was added to precipitate residual PEG. Following an incubation period of 10 minutes on ice, the inoculum was centrifuged at 12,000 g, and the clarified virus containing supernatant was stored at 4ºC until further use. Virus titration was conducted using end-point dilution, and the concentration was given as tissue culture infectious dose 50 (TCID50)/ml. 8 The final inoculum volume was 0.05 ml.
Experimental design
The LD50 for each of the eight virus strains was determined by using an optimised between-patient three-level RSP design, 11 with three design levels using virus doses calculated from the results in the previous level (Table 2). If the calculated dose equalled 0 for any of the virus strains, a minimum dose of 0.1 log10 TCID50 was used. As the previous level functions as a control group for the next level, the need for controls are implemented in the design, and traditional control groups are not necessary. To calculate LD50 for one virus strain, an experimental group of 15 (3 + 5 + 7) mice was used, and a total of eight experimental groups were included (one for each virus strain). The experimental unit was each individual animal, and the experimental end point was 21 days post infection.
Formulae for calculation of dose values (log10 TCID50) in an optimised three-level response surface pathway design.
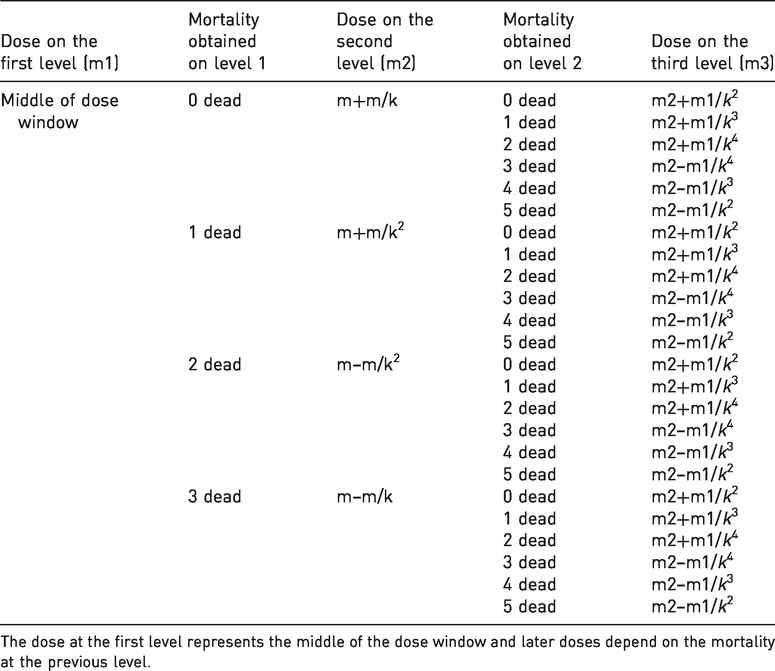
The dose at the first level represents the middle of the dose window and later doses depend on the mortality at the previous level.
The initial dose at level 1 was the middle of the dose window, which had an upper limit that equalled the maximum titre of each individual virus strain. The dose adjustment k-factor was calculated, as described by Holand et al.15,16 The outcome variable was survivor and dead in each of the three levels.
In order to avoid cage and litter variables, the allocation of mice to different virus strains was done by complete randomisation. 25 The study was further performed as observer blind, as the veterinarian who inoculated the mice and performed the clinical assessments did not know which animal was inoculated with which virus.
The criteria for the experimental outcome were as follows
Animals that were encountered dead or cannibalised the first 24 hours after inoculation were recorded as having died from non-specific causes and were excluded from the analysis. Animals that were encountered dead, euthanised due to having reached a humane end point or were cannibalised between 24 hours and 21 days post inoculation were counted as dead. Animals that survived to euthanasia at 21 days post inoculation were counted as survivors.
For each virus strain, cumulative mortality data from the three design levels were finally used to construct a dose–response curve for the virus test strains. 26
Inoculation procedure
Mice were inoculated intracerebrally at 3 days of age. Prior to inoculation, they were given pre-emptive analgesic (Temgesic, buprenorphine, 0.025 mg/kg subcutaneously) and anaesthetised with an inhalation anaesthetic (isoflurane). Bench surfaces were cleaned and disinfected with 70% alcohol, and the mice transferred from their cage to the tray on the working bench. One mouse at a time was carefully physically restrained and prepared for inoculation. The inoculation site was wiped with alcohol. The inoculum (0.05 ml) was carefully injected into the frontal lobe of the left cerebral hemisphere using syringes with 31 gauge needles as a sterile procedure before the inoculation site was wiped again with an alcohol wipe to disinfect any leakage of the inoculum after the injection, and the animal was placed back in the cage. Following inoculation, mice were observed for one hour to detect potential acute toxicity or anaphylaxis.
Statistical analysis
The LD50 results for each experimental group are expressed with 95% confidence intervals (CI) using isotonic regression 26 and trimmed Spearman–Karber regression estimation. 27 The isotonic regression was performed using the pooled adjacent-violators algorithm. 26 The CIs were obtained using parametric bootstrap numerical methods. 28 The trimmed Spearman–Karber program originated from Montana State University and was modified at the Duluth and Athens National Exposure Research Laboratories. The LD50 in the Organisation of Economic Cooperation and Development’s up-and-down procedure was analysed by maximum likelihood estimation using AOT425 software. 29
Results
The mortality on each design level and the final estimates of LD50 for each experimental group are illustrated in Figures 1–3 and presented in Table 3. Two mice died within 24 hours post infection and were excluded from the analysis. Out of the 118 mice, 85 were encountered dead; 64 were found dead, of which 28 were either fully (n = 19) or partially (n = 9) cannibalised, and 21 were euthanised due to reaching a humane end point before the end of the experimental period. The median number of days until death/euthanasia was 9 days. Clinical signs included failure to nurse, ruffled fur and/or neurological signs. The number of diseased/dead in each design level is also shown in Table 3.
Estimated lethal dose 50 (LD50) and ranking of parental (P) and reassortant (R) strains based on cumulative mortality in the optimised between-patient three-level RSP design.
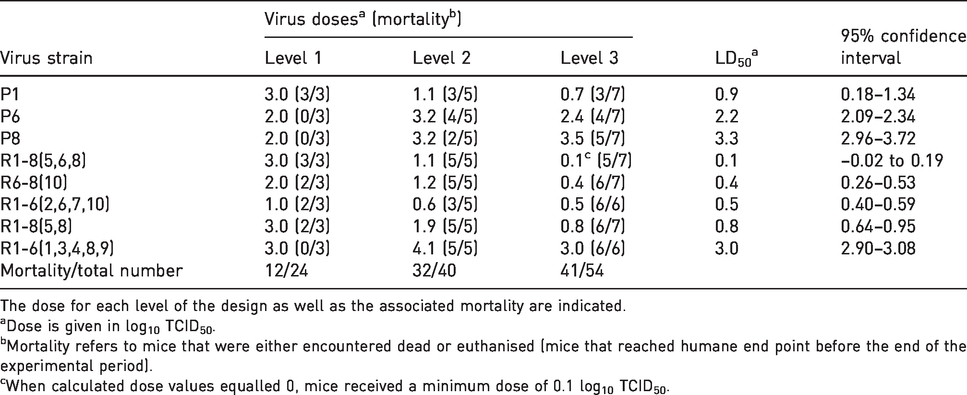
The dose for each level of the design as well as the associated mortality are indicated.
aDose is given in log10 TCID50.
bMortality refers to mice that were either encountered dead or euthanised (mice that reached humane end point before the end of the experimental period).
cWhen calculated dose values equalled 0, mice received a minimum dose of 0.1 log10 TCID50.
Results in each level of an optimised three-level response surface pathway design for three parental virus strains in an in vivo animal experiment in a murine model. The name of the virus strain (P1, P6 and P8) is shown below. The numbers in huddles show number of euthanised or dead mice out of numbers tested.
Results in each level of an optimised three-level response surface pathway design for three reassortant virus strains in an in vivo animal experiment in a murine model. The name of the virus strains is shown below. The numbers in huddles show number of euthanised or dead mice out of numbers tested.
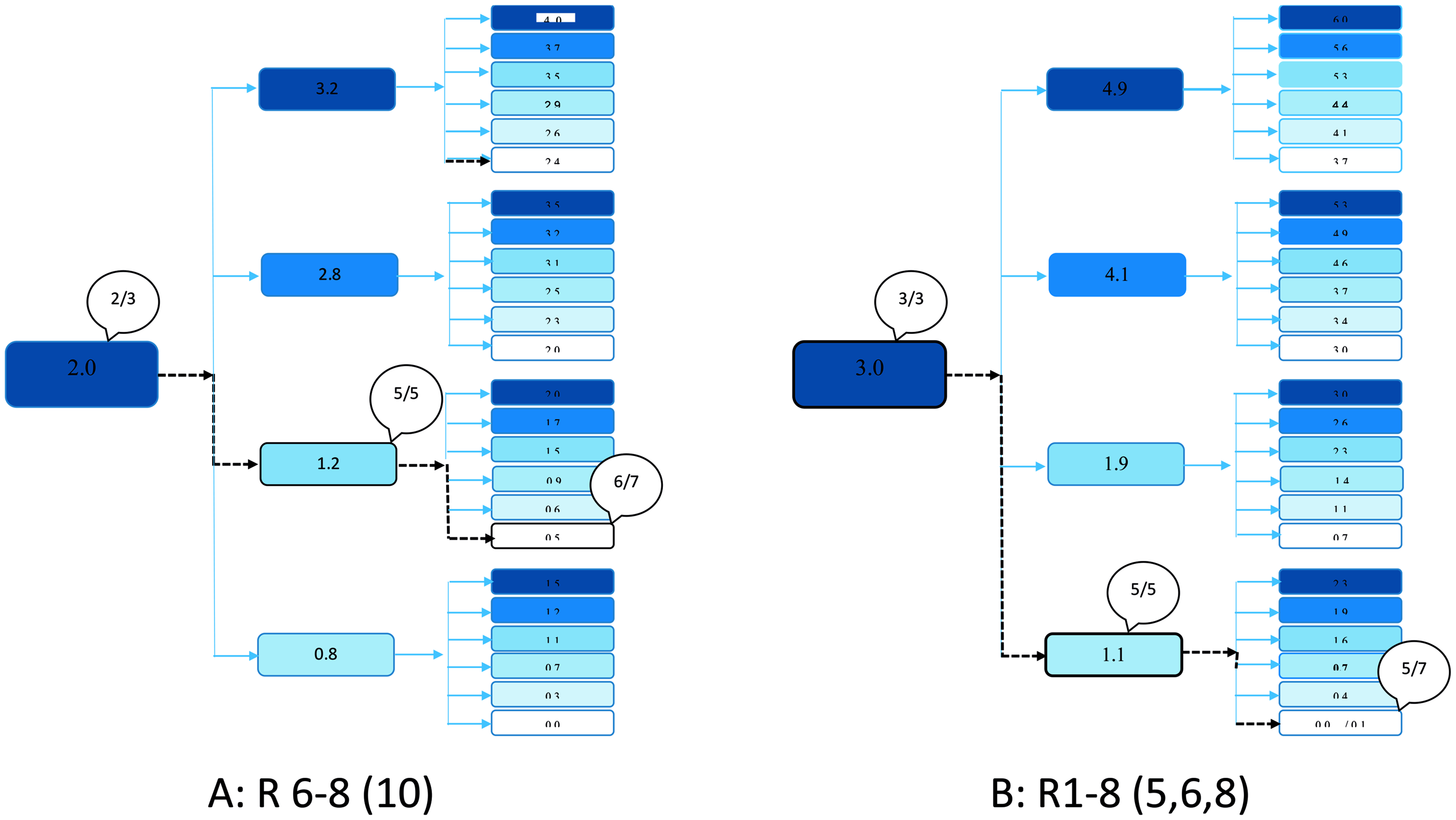
Results in each level of an optimised three-level response surface pathway design for two reassortant virus strains in an in vivo animal experiment in a murine model. The name of the virus strains is shown below. The numbers in huddles show number of euthanised or dead mice out of numbers tested.
The estimated LD50 with 95% CI for each virus strain is shown in Table 3. The LD50 varied from 0.1 to 3.3 TCID50/ml, and the three parental strains differed significantly, as the 95% CIs did not overlap. The lowest LD50 was found for P8 and the highest for P1.
Only one of the reassortants showed lower virulence than both the parental virus strains: reassortant strain R1-6(1,3,4,8,9). The other BTV1-6 reassortant, R1-6(2,6,7,10), showed higher virulence (lower LD50) than both parental strains. The mono-reassortant tested, R6-8(10), also demonstrated a significantly lower LD50 compared with the parental strains. Of the two BTV1-8 reassortants studied, R1-8(5,8) showed little change in LD50, while R1-8(5,6,8) gave significantly lower LD50.
Discussion
In the present study, three parental and five reassortant BTV strains were ranked by LD50 in mice using a refined experimental design. The virus strains had earlier been characterised in regard to phenotype properties in mammalian cell cultures, including viral replication kinetics, the rate for induction of cytopathogenic effect, the effect on cell viability and mechanisms of cell death (apoptosis/necrosis). 18 In general, the results from these cell culture assays and the present study showed little agreement for both the parental virus and the reassortant strains. The basis for this is not known, but most likely it reflects the limitation in the applicability of cell culture models to predict in vivo virulence.
The two parental virus strains P1 and P6 were derived from attenuated virus strains. However, both showed higher virulence in mice compared with parental strain P8 which derived from BTV8 and were known to be highly virulent in ruminants in the field. 30 This indicates that despite an improved design, the gap between findings in mice and the outcome in ruminants is considerable and unpredictable. The clinical outcome in ruminants is a multi-factorial phenomenon influenced not only by intrinsic differenced in the BTV phenotype but also by external factors such as host, vector and environment.
The results of the present study show that at least in a mice model, reassortant BTV can have altered virulence properties compared with their parental strains. For African horse sickness, which is caused by a closely related Orbivirus, outbreaks of disease were confirmed to have been caused by reassortant viruses with genome segments derived from live attenuated vaccine strains. 31 Reassortant BTV strains have also been found in cattle, and in vivo co-infections with BTV vaccine strains have demonstrated genetic reassortment.32,33 Ruminants might provide a suitable host for BTV reassortment, with production of reassortants that spread in the population with subsequent negative consequences.
Only one of the reassortants in the present study, R1-6(1,3,4,8,9), showed lower virulence than both parental virus strains. This may indicate that no genome segment 1,3,4,8 or 9 from P6 is associated with increased virulence. Since the inverse BTV1-6 reassortant, R1-6(2,6,7,10), showed the lowest LD50, it might indicate that segment 2,6,7 and/or 10 from P6 caused increased virulence when combined with the segments from P1. In addition, the only mono-reassortant tested, R6-8(10), further support segment 10 as virulence-causing, since it significantly lowered LD50 compared with the parental strains, at least in combination with the P6 segments. Segment 10 codes for a non-structural protein NS3/NS3A, which is indispensable for viral release in cells. 17 From the BTV1-8 reassortants studied, it seems that receiving segment 5 and 8 from the supposed more virulent BTV-8 has little influence on virulence, while receiving segment 5,6 and 8 from BTV-8 appeared to cause significant lower LD50. This could indicate that segment 6 also causes higher virulence. More information on the genome segments that contain BTV virulence markers and their combinations is required, although investigations have been performed in both mice and ruminants.4,34,35 Generation of sequential mono-reassortants through reverse genetics has been attempted, 5 and this approach followed by phenotype analysis may narrow down the genome segments to study in order to find mutations and genomic features responsible for influencing virulence.
Using live animals for the study of pathogen virulence is ethically highly challenging. The traditional study design may have contributed to the unnecessary use of many animals but may also result in suboptimal statistical power and research quality. This may contribute to the general problem with replication of results from live animal experiments. In the field of virology, total replacement of live animal experiments might not be possible. However, the two other factors in the 3R concept – reduction and refinement – should be achievable. The fact that 15 mice used to establish the LD50 of each virus strain in the present study using the RSP design represents a significant reduction in the number of required animals compared with the traditional designs. The rapid converge towards the dose of interest and the implementation of controls in the design represent other benefits which reduce the necessary number of animals and increase the statistical power.11,12,16
Standardisation of production of inoculum is a general challenge in LD50 studies in virology, since viruses requires a biological system (cells) to multiply. This may be one of the reasons for lack of uniformity in viral LD50 studies, which makes both in vitro and in vivo virulence data difficult to compare and reproduce between laboratories. Different virus passage histories and titration methods might be used and may be difficult to standardise between research groups. The choice of virus titration method could also influence the accuracy of the concentration estimate – the statistical TCID50 method compared with viral count by the plaque forming unit method. Potential toxicity in the inoculum itself was considered in the present study, particularly with the PEG6000 method used to concentrate virus from cell culture supernatant due to high salt concentrations. Any toxic effect was, however, discarded as the majority of mice survived for more than 24 hours post inoculation.
It is generally recommended that adult mice are acclimatised for three to 7 days prior to initiation of experimental work. 36 In the present study, practical reasons and the short susceptibility period for mice to BTV prevented a longer acclimatisation period than overnight. Even if the animals had been procured as pregnant dams, the requirement that all pups be synchronised to 3 days (from an experimental/logistical perspective) would have been difficult to achieve. Several factors may have contributed to stress for dams, such as transport, monitoring and handling of offspring. These stressors may have affected their behaviour, which again could negatively affect the survival of pups and result in abnormal behaviours such as cannibalism. In murine experiments, the litter is traditionally the experimental group. In the present study, complete randomisation of the test strains independently of litter or cage was done to avoid litter bias. However, litter bias might still be present, as mortality in the litter may conversely have caused behavioural changes in the dams that could have influenced pup survival. The randomisation also made it necessary to label the pups. Different labelling methods were tested, but no procedures were found to represent a reliable and practical alternative to toe clipping of the newborn mice. This method is discouraged and prohibited in several European countries but legal in South Africa. The method is discussed in a report of the Federation of European Laboratory Animal Science Associations working group, where it is emphasised that toe clipping should only be considered at an early age, before the ossification process is completed and when it is most difficult to find other options. 37 For further use, a reliable but less invasive and stressful labelling technique should be sought.
Sex is also a factor that might influence the results of a live animal experiment. The WHO generally recommends using the same number of males and females for rodents older than 6 weeks of age, and differences in susceptibility of adult male and female mice have been observed for several infections. For virulence studies in new-borns, on the other hand, it is common to use whole litters.4,38,39 For several infections that cause encephalitis in mice, the different susceptibility has been related to sexual maturity. 40
In the present study, death was used as end point, which is not optimal and ethically challenging. BT infection in newborn mice has earlier been documented to lead to rapid development of acute, fatal encephalitis.41,42 This rapid disease development makes it difficult to find an earlier suitable end point that could be monitored in these young animals. Both neurological changes and failure to nurse, which included all clinical signs observed, were defined as humane end points, and the animals were euthanised. An animal model with a slower development of disease could make it possible to find a more sophisticated end point.
The handling, labelling and inoculation procedure with anaesthetisation are also considerable stress factors for newborn mice and might have contributed to non-specific mortality both directly and through stress to the dams. This may have influenced the LD50 estimates as such, but due to the randomisation procedure, the level of non-specific mortality can be assumed to be constant between experimental groups, and the ranking of the strains should still be valid.
The use of adult IFNAR(–/–) mice combined with a three-level RSP design may be a suitable design for future BTV virulence studies. Although these animals are more expensive, less available and require specialised isolation units, use of such mice would allow an ethically and scientifically better design that could address some of the challenges encountered.
The present study generated data that supported that reassortant BTV strains may have altered in vivo properties compared with the parental strains. It should, however, be emphasised that extrapolation of BTV virulence results between cell culture, mice, and ruminant models is challenging. Further improvement of LD50 studies in virology will require a multidisciplinary approach between virologists, animal laboratory experts and statisticians, so that potential difficulties in the study design, the experimental procedures and animal model can be addressed. The optimised three-level RSP design for viral LD50 studies should be strongly considered from animal welfare, cost and scientific perspectives.
Footnotes
Acknowledgements
The authors would like to express their gratitude to ARC-LNR for allowing us the use of the BSL-3 Transboundary Animal Diseases Facility, and to the staff there for assistance with the mice experiments. We would also like to thank Dr P. Van Rijn and Dr R. Van Gennip who kindly provided all virus strains, Dr Sagita Dewi for assistance with data analysis and Dr Siri Kristine Knudsen for fruitful discussions and critical review of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: The project was financially supported by the Norwegian University of Life Sciences (NMBU), TINE SA (cooperative dairy organisation, Norway), Animalia (Norwegian Meat and Poultry Research Centre), The South African National Research Foundation and the University of Pretoria (South Africa).