
Abstract
We have previously demonstrated that purified virgin mouse mammary luminal epithelial and myoepithelial cells promiscuously express cell type-specific cytokeratins when they are cloned in vitro. Changes in cytokeratin expression may be indicators of the loss or change of the differentiated identity of a cell. To investigate the factors that may be responsible for the maintenance of differentiated cellular identity, specifically cell-cell and cell-matrix interactions, we cloned flow-sorted mouse mammary epithelial cells on the extracellular matrix (ECM) derived from the Engelbreth-Holm-Swarm murine sarcoma (EHS matrix). Changes in cell differentiation on EHS, compared with culture on glass, were analyzed by comparing patterns of cytokeratin expression. The results indicate that ECM is responsible for maintenance of the differentiated identity of basal/myoepithelial cells and prevents the inappropriate expression of luminal antigens seen on glass or plastic. Luminal cell identity in the form of retention of luminal markers and absence of basal/myoepithelial antigens, on the contrary, appears to depend on homotypic cell-cell contacts and interactions. The results also show that luminal cells (or a subpopulation of them) can generate a cell layer that expresses only basal cytokeratin markers (and no luminal cytokeratin markers) and may form a pluripotent compartment.
We have previously described (Smalley et al. 1998) a system for the separation of virgin mouse mammary epithelium by flow cytometry and methods for the clonal culture of such separated cells on tissue culture plastic and glass. We have characterized mouse mammary luminal epithelial and myoepithelial cell-derived clones on the basis of phase-contrast morphology and cytoskeletal marker expression. Flow-sorted mouse mammary luminal epithelial cells gave rise to three morphologically identifiable clone types in vitro, whereas myoepithelial cells gave rise to only one. We also demonstrated that the expression of cell type-specific cytoskeletal markers is deregulated in vitro, leading to promiscuous expression in luminal epithelial clones of basal/myoepithelial markers and vice versa. Deregulation of these cytokeratin markers is never seen in cloned human mammary epithelial cells in vitro (O'Hare et al. 1991) and is seen only to a limited extent in a subpopulation of cloned rat mammary luminal cells in vitro (Dundas et al. 1991).
Cytokeratins are important in tissue architecture and cell organization in vivo (Vassar et al. 1991), mediating aspects not only of cell identity but also cell polarity (Parry et al. 1990). Expression of different cytokeratins has furthermore been shown to be diagnostic of mammary epithelial cell types in vivo in human (Taylor-Papadimitriou and Lane 1987) and rat (Dundas et al. 1991) and also of luminal epithelial and myoepithelial cells in vitro in both species (Dundas et al. 1991; O'Hare et al. 1991). The deregulation and promiscuous expression of cytokeratin markers in mouse mammary epithelial cells in vitro therefore suggests not only that these cells lose some aspect of their basic cell identity when put into culture but also that the cell identity and differentiated state of virgin mouse mammary epithelial cells is not cell-autonomous but requires constant extracellular signals. These signals are not provided for in separated cells grown on tissue culture plastic or glass, and this may be why they begin to lose their basic luminal epithelial/myoepithelial identity.
There are at least two possibilities for the extracellular signals that could reinforce cellular identity in vivo. First, each cell type could require contact with, or paracrine signals from, other cells, either of the same type or of different types. Thus, myoepithelial cells might require signals from luminal cells, and vice versa. When put in separated cultures and lacking signals from the other cell type, the cells could then begin to dedifferentiate. The second possibility is that mouse cells are especially sensitive to the presence or absence of extracellular matrix (ECM). ECM not only provides a support for cells in vivo but its components also act as ligands that interact with cells via cell surface molecules which include the integrins (Streuli et al. 1991; Hynes 1992; Streuli 1993; Juliano 1994) and cell surface β-1,4-galactosyltransferase (Barcellos-Hoff 1992). ECM is required for the morphological and functional differentiation of a number of cell types, including mouse mammary epithelial cells (Li et al. 1987; Blum and Wicha 1988; Streuli 1993). Indeed, single mouse mammary cells derived from midpregnant mice suspended in the ECM derived from the Engelbreth-Holm-Swarm murine sarcoma (EHS matrix) (Kleinman et al. 1984) will maintain β-casein synthesis although they do not proliferate (Streuli et al. 1991), and ECM has also been shown to be a requirement for the suppression of apoptosis in mammary epithelium (Pullen et al. 1996). Disruption of β1-integrin function in the mammary epithelium of transgenic mice has been shown to perturb mammary epithelial proliferation during pregnancy (Faraldo et al. 1998), and modulation of integrins by use of blocking antibodies has been demonstrated to restore some aspects of functional normality to early stages in malignant progression of breast cells (Weaver et al. 1997).
To test the hypothesis that maintenance of the differentiated identity of mouse mammary epithelial cells requires ECM, we cultured separated virgin mouse mammary luminal epithelial and myoepithelial cells on EHS matrix at clonal density. The morphology of the resulting clones was examined and the expression of cytokeratins was used as an index of whether the in vivo differentiated identity of the isolated cells was maintained by culture on the EHS matrix. The results show that, in the presence of ECM, mouse mammary myoepithelial cells formed flat clones that maintained a more differentiated phenotype, with a marked reduction in the extent to which inappropriate antigens were expressed. Mouse mammary luminal epithelial cells under similar conditions formed either flat clones, some of which generated a layer of cells with basal/myoepithelial characteristics, or spheroidal clones (“mammospheres”) in which the central mass of cells, which had no contact with ECM but were surrounded in all directions by other cells of luminal origin, retained a more differentiated phenotype and did not express basal markers. The outer layer of mammospheres still expressed both luminal and basal markers and interestingly it was only this outer cell layer which produced β-casein in response to lactogenic hormones.
These results demonstrate that the maintenance of luminal phenotype in mouse mammary cells is likely to be a function of homotypic cell-cell interactions, whereas ECM signaling is likely to be primarily responsible for the maintenance (and generation) of the basal/myoepithelial phenotype, although it also appears to be a requirement for the functional differentiation of luminal cells. The results also provide evidence for the existence of a pluripotent compartment within the adult mouse mammary luminal epithelium.
Materials and Methods
Isolation of Mouse Mammary Epithelial Cells
This has previously been described in detail (Smalley et al. 1998). In brief, mixed populations of virgin mouse luminal epithelial and myoepithelial cells were isolated from the fourth mammary fat pads of 10-week-old virgin female Parkes mice (Medical Research Council) by fine mechanical mincing followed by collagenase digestion to liberate organoids. After washing, most of the stromal cells contaminating the preparation were isolated by a differential attachment procedure, after which the organoids were digested using polyvinyl pyrrolidine-EGTA-trypsin (PET) Cell Dissociation Solution (Techgen International; London, UK). The resulting suspension consisted mainly of single epithelial cells, with some contaminating stromal cells as well as some debris (including blood vessel fragments) and residual multicellular epithelial clumps. These were removed by filtering the cell suspension through a 35-μm nylon mesh (Lockertex; Warrington, UK).
Immunofluorescence Staining of Cell Surface Antigens for Flow Cytometry
Mouse mammary luminal epithelial or myoepithelial cells were isolated from the prepared cell suspension by flow cytometry, as previously described (Smalley et al. 1998). The cells were split into two samples, one of which was stained with a cell type-specific antibody and the other of which was kept as a no first antibody (NFA) control. Both samples were incubated with a Serotec (Kidlington, UK) FITC-conjugated F(ab')2 rabbit anti-rat IgG antibody and the sample labeled with the primary antibody was then sorted into labeled and unlabeled cells, with the NFA control sample being used to correct for nonspecific fluorescence.
Cell type-specific antibodies used were rat monoclonal antibodies (MAbs) obtained from Dr. A. Sonnenberg. MAb 33A10, which is specific for a milk fat globule membrane antigen, was used to specifically isolate luminal epithelial cells. MAb JB6, which in our hands uniformly stains the basal/myoepithelial layer of the mammary epithelium in Parkes mice (Smalley et al. 1998), was used to isolate this cell layer. The antigen recognized by this antibody has not, to our knowledge, been fully characterized at this time. In BALB/c mice this antibody stains only basal cells and not differentiated myoepithelial cells (Sonnenberg et al. 1986). The difference between our results and the results of Sonnenberg et al. is not clear; we can only suggest strain differences. It is known that Parkes mice and BALB/c mice reach different stages of differentiation in the virgin gland (i.e., Parkes mice show a greater degree of lobuloalveolar differentiation in older virgin animals), and these strain differences may also occur at the cellular level.
Flow Cytometry
Flow cytometry was used for separation of cell samples stained with antibodies against cell surface antigens. This enabled the isolation of purified individual epithelial cells that were either positive or negative for a particular surface antigen, because they were first analyzed and gated as single cells on the basis of forward and orthogonal light scatter before identification and separation of fluorescent cells.
An Ortho 50H Cytofluorograf cell sorter equipped with a 2150 data analysis system and an argon ion laser tuned to give 50 mW at a wavelength of 488 nm was used. Recorded parameters were light-scattered orthogonally and at a narrow forward angle at 488 nm and green fluorescence at 520 nm [from fluorescein isothiocyanate (FITC) excited at 488 nm]. Cells were sorted from a flow stream of sterile PBS, and using anti-coincidence circuitry one drop (containing one cell) was deflected for each sort command.
Mammary Epithelial Cell Cloning
Primary mouse mammary epithelial clone cultures were established on ethanol-sterilized 13-mm glass coverslips in Nunc (Roskilde, Denmark) 24-well plates. Coverslips were either left uncoated or were coated with 250 μl of EHS murine sarcoma matrix (Collaborative Research; Cambridge, UK). This was allowed to set into a thick gel by incubation at 37C for 1 h before medium was added to the wells.
Flow-sorted mouse mammary luminal epithelial and myoepithelial cells were cultured using a feeder layer of irradiated (20 Gy from a 60 Co source) 3T3-L1 preadipocyte cells at a density of 20 cells mm−2 and a growth medium consisting of a 1:1 mixture of DMEM and Ham's F12 with 10% FCS, 5 μl−1 insulin, and 10 ng ml−1 cholera toxin. Freshly disaggregated mouse epithelial cells were plated at a density of 2.5 cells mm−2 (equal to approximately 500 cells per well) and cultures were maintained at 37C in an atmosphere consisting of 5% (v/v) oxygen, 5% (v/v) CO2, and 90% (v/v) nitrogen. These growth conditions had previously been determined as optimal for clonal culture of virgin mouse mammary epithelium (Smalley et al. 1998). Overall cloning efficiencies on EHS-coated coverslips and glass or uncoated plastic were comparable (± 10%).
To examine functional differentiation, clones grown on glass or on EHS matrix were cultured in the basic growth medium or in medium containing ovine prolactin at 5 μg/ml and hydrocortisone at 1 μml (“lactation medium”; Streuli et al. 1991). After 8 days, cultures were fixed and stained for β-casein.
Immunofluorescence Staining of Epithelial Clones In Vitro
After 8 days in culture, epithelial colonies were observed, fixed and stained. Table 1 gives a list of mouse MAbs used for staining of mouse mammary cells in vitro. LE61, LE65, LLOO2, and LP2K were obtained from Professor E.B. Lane (Lane 1982; Lane et al. 1985; Purkis et al. 1990) and RCK107 was obtained from Professor F. Ramaekers (Wetzels et al. 1991). The mouse MAb 1A4 against α-isoform smooth muscle actin was obtained from Sigma (Poole, Dorset, UK). RβC-1, a mouse anti-rat β-casein MAb that cross-reacts with mouse casein, was obtained from Dr. C. Kaetzel (Kaetzel and Ray 1984).
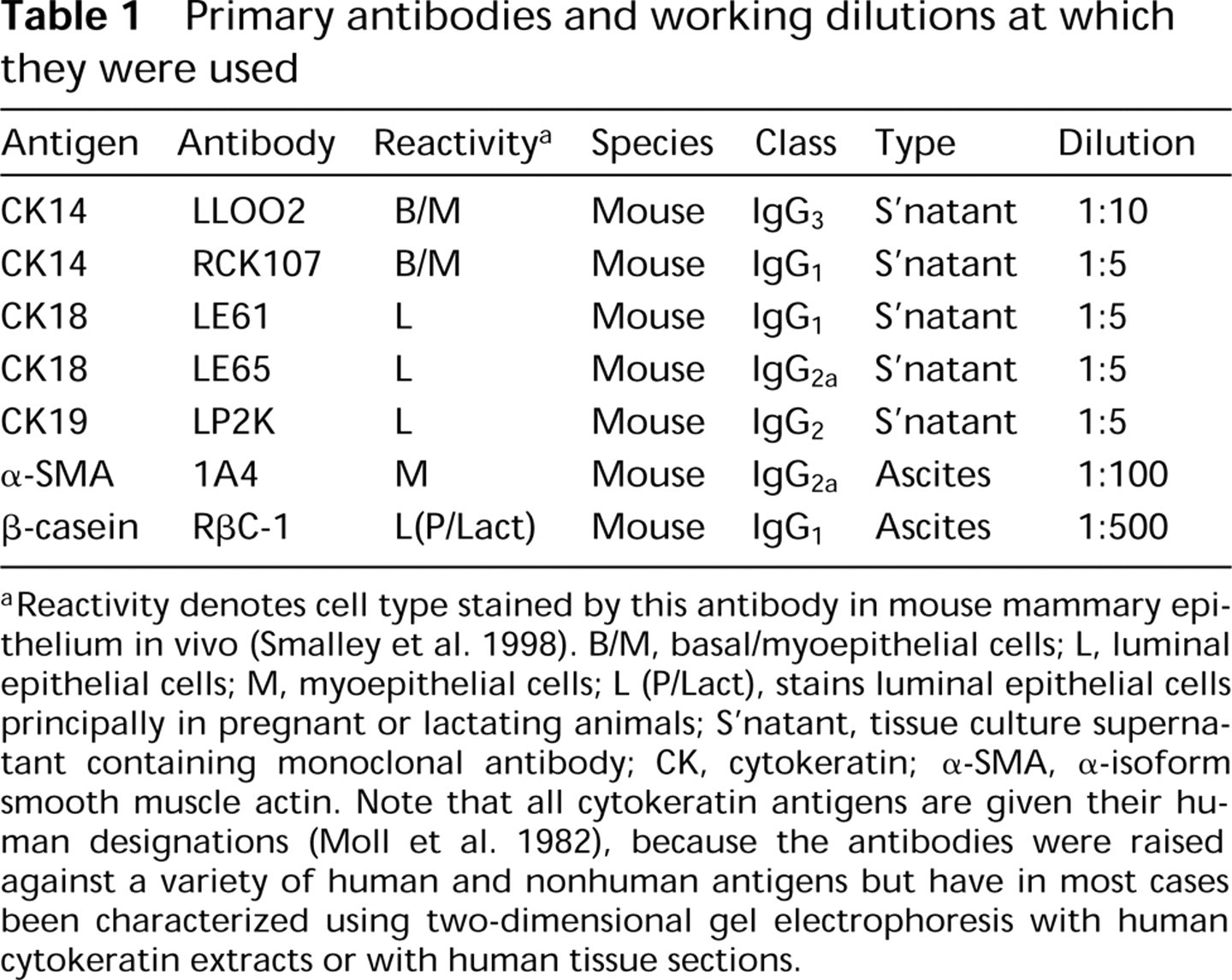
Primary antibodies and working dilutions at which they were used
Clones on glass coverslips were methanol-fixed and stained in the wells of the 24-well plates in which they had been grown, as previously described (Smalley et al. 1998). In brief, coverslips were incubated with a mixture of one, two, or three primary mouse MAbs at the appropriate dilution, followed by washing and then incubation with a mixture of diluted class- and subclass-specific second antibodies. These were conjugated to one of three fluorochromes (amino methylcoumarin acetate, AMCA; fluorescein isothiocyanate, FITC; Texas Red, TX). Coverslips were washed a second time and then mounted in Citifluor AF1 anti-fadent (Citifluor; Canterbury, Kent, UK) and examined. Preliminary studies using combinations of single, double, and triple labeling demonstrated no difference in staining patterns in single compared to multiple stains. There was no evidence for competition between antibodies.
Phase-contrast and Fluorescence Microscopy
A Zeiss Axiovert 100 infinity-corrected microscope fitted with a Zeiss MC80 photomicrograph apparatus was used for phase-contrast and fluorescence microscopy. A mercury vapor lamp was used for epifluorescent illumination and the microscope was also fitted with a triple filter block apparatus containing wedge-free emission filters for FITC (Zeiss filter set 10) and TX (Zeiss filter set 00, also used for TRITC) as well as a filter for AMCA (Zeiss filter set 02). So that AMCA fluorescence could be observed and photographed in multiply stained samples without breakthrough from the FITC or TX/TRITC, the microscope was also fitted with an extra emission filter (transmission cut-off at 500 nm) for use with AMCA only.
The majority of phase-contrast images were photographically recorded using a × 10 Achrostigmat (NA 0.25) objective. Fluorescence images were recorded using a × 25 water immersion Neofluar (NA 0.80) objective. Phase-contrast photomicrographs were taken on Kodak Technical Pan 35-mm film (100 ASA). Black-and-white fluorescence photomicrographs were taken using Kodak T-Max Professional P3200 35-mm film.
Confocal Microscopy
For observation of certain samples stained by immunofluorescence, a Bio-Rad MRC 600 confocal imaging system equipped with a krypton-argon laser was used in conjunction with a Nikon Optiphot fluorescence microscope. Samples were observed using a × 60 plan Apo objective lens.
Scanning Electron Microscopy
Coverslips for scanning electron microscopy (SEM) were prepared by standard protocols (Kay 1965) and then critical point-dried in a Polaron Critical Point Drier. Coverslips were mounted, sputter-coated using a gold/palladium mixture, and observed in a Hitachi S800 scanning electron microscope.
Results
Myoepithelium-derived Clones: Morphology
Ten-week-old virgin Parkes mouse mammary myoepithelial cells separated by flow cytometry using MAb JB6 were plated at clonal density onto either uncoated glass coverslips or coverslips coated with EHS gel and were observed after 8 days in culture. Morphological comparisons between myoepithelium-derived clones on uncoated and EHS-coated coverslips were made on clones stained by multiple immunofluorescence for cytoskeletal markers, because clones cultured on EHS were difficult to observe directly by phase-contrast microscopy. This was a result of the flattened nature of the cells comprising these clones, the granular background of the underlying matrix, and optical limits of phase-contrast microscopy in the wells of 24-well plates, especially at the edges of such wells.
The majority of clones that formed (>90%) on EHS matrix had a very similar morphology, being monolayers composed of large, flat cells (Figure 1C and 1D), similar in appearance to myoepithelium-derived clones on tissue culture plastic or glass (Figure 1A and 1B) (Smalley et al. 1998). However, in colonies on EHS gel the cells were often more closely packed together than those on glass, giving the colonies a “paving-stone” appearance. There was little variation in the appearance of this clone type even when the clones were growing on the thick layer of gel that formed at the edges of the coverslips (as opposed to the thinner coating at the center). A minority of clones (<10%) consisted only of small balls of cells with no monolayer component, which appeared to be embedded in the matrix (not shown). Apart from an increase in the size of clones, no morphological changes were observed in the clones after periods of culture longer than 8 days.
Myoepithelium-derived Clones: Staining for Cytoskeletal Antigens
All cells of all myoepithelium-derived clones on EHS gel were strongly stained by an antibody (LLOO2) to cytokeratin 14 but only approximately 10% or fewer of the cells in such clones reacted with LE61, an anti-cytokeratin 18 antibody, as demonstrated by double indirect immunofluorescence (Figure 1C and 1D). This was in contrast to the myoepithelial clones on glass, which showed similar levels of cytokeratin 14 reactivity but a much higher proportion of cytokeratin 18-positive cells (about 50%), as we have previously described (Figure 1A and 1B) (Smalley et al. 1998).
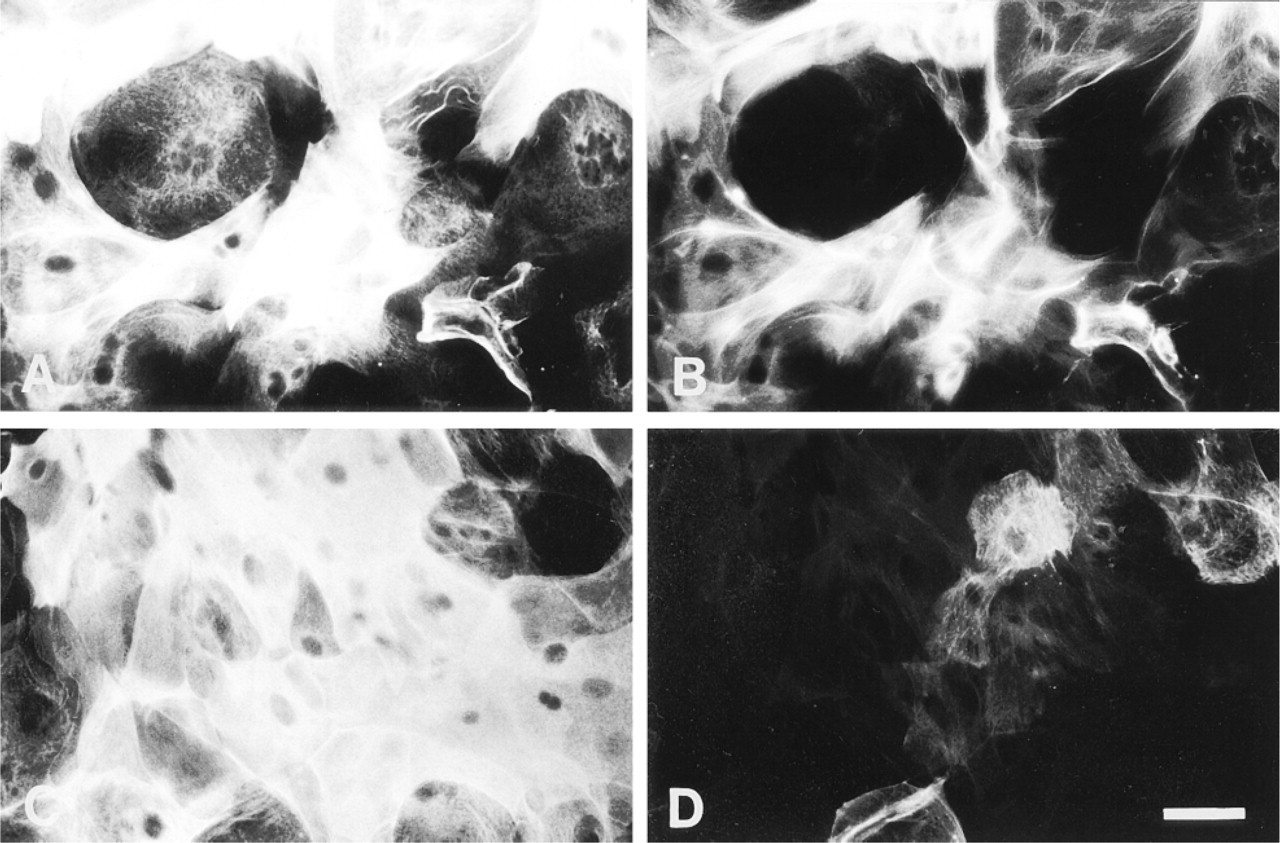
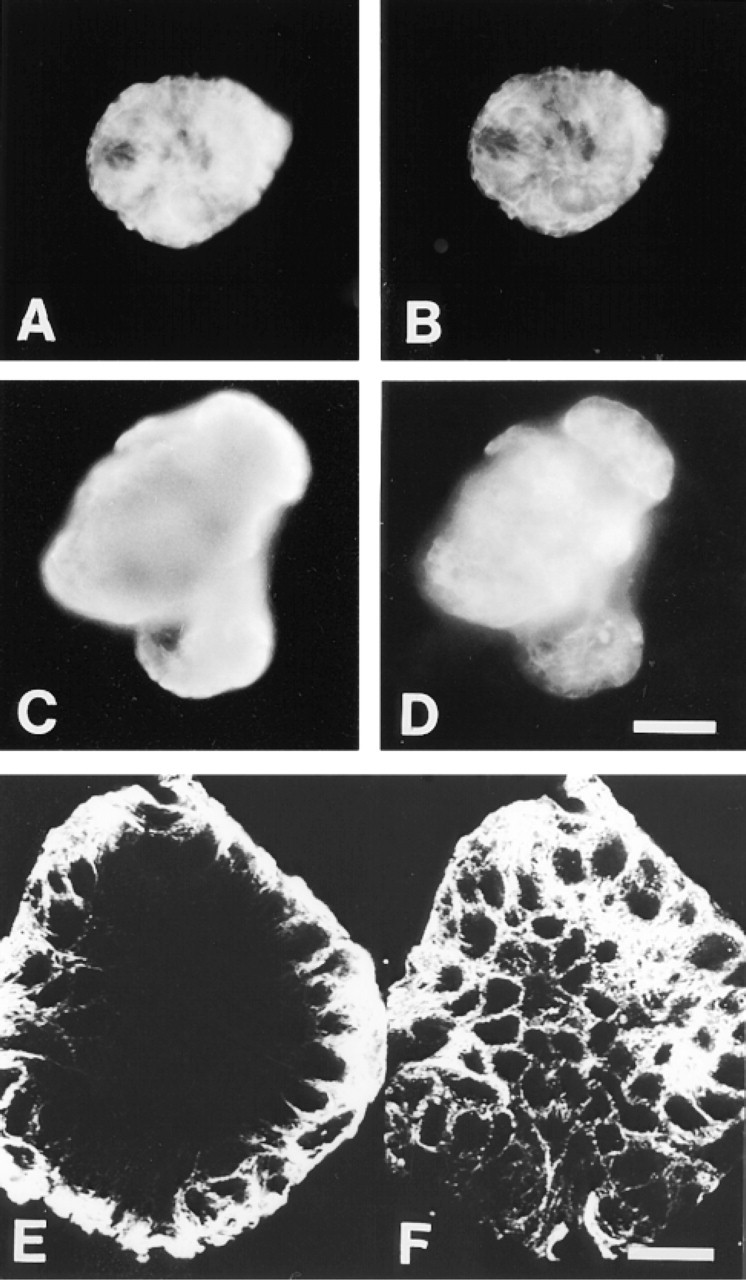
Double staining of mouse mammary myoepithelial clones with LLOO2 (cytokeratin 14; A,C) and LE61 (cytokeratin 18; B,D). (A,B) Clone cultured on uncoated glass. (C,D) Clone cultured on EHS matrix. Bar = 30 μm.
The strictly basal nature of the myoepithelium-derived clones on EHS gel was confirmed by triple indirect immunofluorescence staining using the antibody combination LLOO2 (anti-cytokeratin 14), LE65 (a second anti-cytokeratin 18 antibody), and RCK107 (a second anti-cytokeratin 14 antibody). All cells in all such clones were strongly stained with both cytokeratin 14 antibodies (LLOO2 and RCK107), whereas none stained with LE65 (not shown). Over 80% of cells also stained for smooth muscle actin (antibody 1A4; not shown), whereas no cells stained for cytokeratin 19 (antibody LP2K; not shown). This staining pattern is characteristic of mouse myoepithelial cells in vivo (Smalley et al. 1998). None of the myoepithelium-derived clone types (i.e., majority flat type or the small minority of rounded balls of cells) showed any evidence of differentiation towards a luminal phenotype in the presence of EHS matrix, as evidenced by their lack of lumen-specific cytokeratins.
Comparable results were obtained from two separate JB6 sorts and 13 separate unsorted cell populations prepared from different batches of mice, each of which was simultaneously plated with and without EHS matrix. Clonal identities were established solely on the basis of sorted cells, and unsorted clones were used merely to increase the numbers of analyzable colonies. Unsorted populations were not used to define clonal identities.
Luminal Epithelium-derived Clones: Morphology
Ten-week-old virgin Parkes mouse mammary luminal epithelial cells separated by flow cytometry were plated at clonal density onto uncoated glass coverslips or coverslips coated with EHS gel at clonal density and observed after 8 days in culture.
After this time, the three types of clone previously described (Smalley et al. 1998) could be seen growing on uncoated glass coverslips. On the EHS gel-coated coverslips, two types of clone morphology could be distinguished by phase-contrast microscopy: flattened clones and rounded but irregular mammospheres. Flat clones were observed in areas where the coating of EHS gel was thinner, i.e., towards the middle of each coverslip, whereas three-dimensional clones grew at the edges where the gel was thickest.
The morphology of the flat clones varied such that at one extreme they consisted of cells forming distinct colonies but with variable morphologies and varying degrees of contact between them, similar to the clones observed on glass (Figure 2A), whereas at the other extreme they were composed of very tightly packed cuboidal/polygonal cells (Figure 2B). The mammospheres were often simple spheroids but were also observed with either long and thin (Figure 2C) or short and club-shaped (Figure 2D) outgrowths, or both. After 8 days of culture, it appeared that the spheroids were partially enveloped by EHS gel and that the club-shaped outgrowths were growing into the surrounding ECM where it was densest. The observation that the spheroids were growing partly surrounded by a layer of the gel rather than on top of the matrix was confirmed by SEM studies. Figure 3 shows a scanning electron micrograph of an 8-day-old mammosphere seen to be growing in cup of fibrous matrix, some of which forms a thin layer extending up and over the sides and top of the sphere.
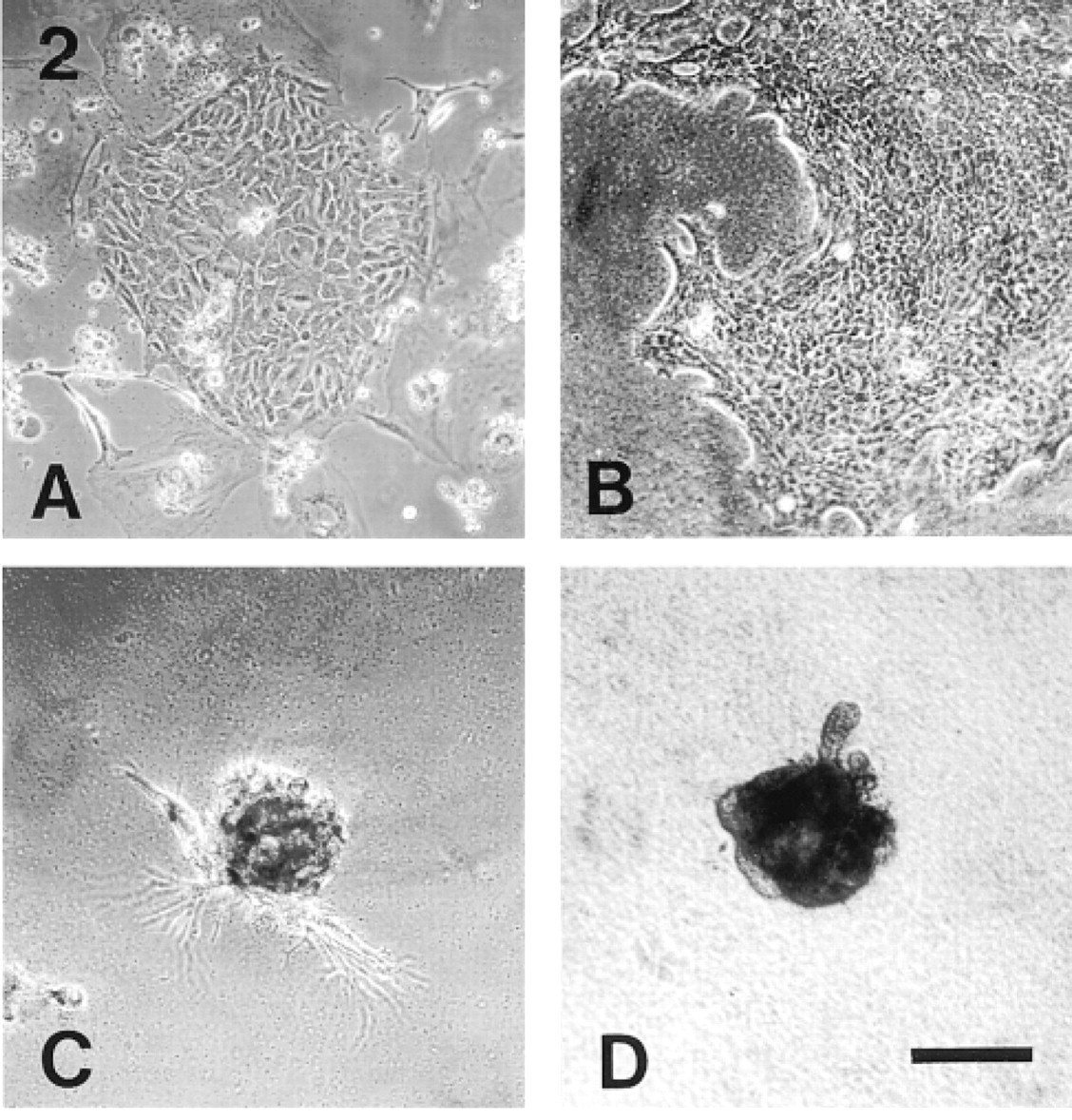
Phase-contrast appearance of mouse mammary luminal epithelial clones. (A) Typical clone cultured on glass. The floating debris is the remains of dead, irradiated feeder cells. (B) Typical monolayer-type clone on EHS matrix gel. (C) Mammosphere with long, thin outgrowths. (D) Mammosphere with club-shaped outgrowths. Bar = 150 μm.
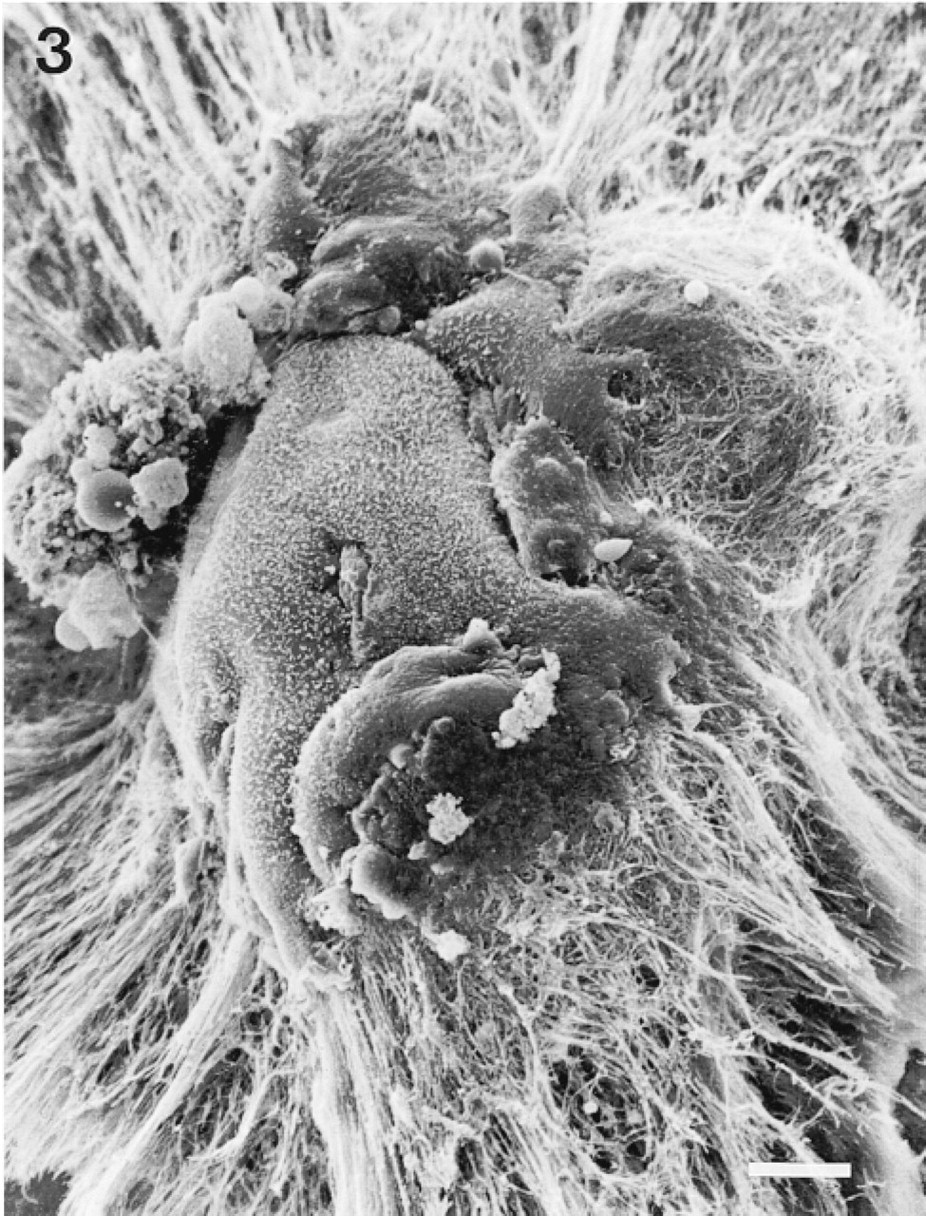
Scanning electron microscopy image of three-dimensional clone on EHS matrix (mammosphere), showing clone embedded in cup of fibrous matrix. Bar = 10 μm.
No further morphological changes were visible under phase-contrast after 8 days in culture, apart from an increase in clone size. The more refractile nature of the cells comprising these clones meant that there was little difficulty in observing them by phase-contrast, especially in the flattened clones composed of cuboidal/polygonal cells and in the three-dimensional clones.
Luminal Epithelium-derived Clones: Staining for Cytoskeletal Antigens
Luminal epithelial colonies grown on uncoated glass coverslips and on coverslips coated with EHS matrix gel were principally characterized by double indirect immunofluorescence staining for cytokeratin 14 (antibody LLOO2) and cytokeratin 18 (antibody LE61).
Clones on uncoated glass coverslips had the same promiscuous staining pattern as previously described (Smalley et al. 1998), i.e., all clones (which were all monolayers as described above) were double stained in > 90% of cells (not shown). The flat clones grown on EHS showed two different staining patterns (Type I monolayer and Type II multilayer). Type I clones were monolayers that appeared very similar to the Type A, B, or C mouse mammary luminal epithelium-derived clones grown on glass (Smalley et al., 1998), with all cells being strongly positive for both cytokeratin 14 and cytokeratin 18 (Figure 4A and 4B). These clones formed approximately 90% of clones seen in the thinner areas of EHS matrix. There was variation within the Type I clones, corresponding to the variation seen in the phase-contrast morphology of the flattened EHS clones, and it was apparent that the cells which formed the clones tended to be more closely packed in areas where they were growing on a distinct layer of matrix, whereas the cells were more spread out (and resembled even more strongly cells cultured on uncoated glass) where little matrix was visible.

Cytokeratin staining of mouse mammary luminal epithelial monolayer clones on EHS matrix. Cultures were double stained with LLOO2 (cytokeratin 14; A,C) and LE61 (cytokeratin 18; B,D). (A,B) Type I clone. (C,D) Type II clone. Bar = 35 μm.
Type II clones formed a small minority of flat clones (approximately 10%) that were seen only on a well-defined matrix layer. However, they were the most interesting because they were multilayered, although this was apparent only after staining (Figure 4C and 4D). Furthermore, the basal layer, in direct contact with the matrix, consisted of cells that were cytokeratin 14-positive but cytokeratin 18-negative. The upper layer consisted of compact cuboidal cells that were a mixture of cytokeratin 14-negative/18-positive as well as cells positive for both antigens.
The emergence of cells within lumen-derived clones that retained a specifically luminal phenotype (i.e., cytokeratin 14-negative/18-positive) was seen even more clearly in the mammospheres, which could be divided into two types on the basis of their cytokeratin staining patterns (Figure 5). The first type consisted of double staining (cytokeratin 14-positive/18-positive) of all the cells of the mammosphere (Figure 5A and 5B). The second type of staining pattern, which was the most common seen in mammospheres (>70%), was typified by a central mass of cells in the colony staining strongly for cytokeratin 18 but not cytokeratin 14, whereas only the outermost cell layer double stained for both cytokeratins 14 and 18 (Figure 5C and 5D). This segregation of phenotypes within mammospheres was clearly seen by confocal microscopy (Figure 5E and 5F). A complete loss of cytokeratin 18 staining in the outer layer of the mammospheres, to give a cytokeratin 14 positive/18 negative layer comparable to the basal layer seen in the Type II (multilayer) lumenderived flat clones was, however, not observed in cultures maintained for up to 8 days.
Lumen-derived clones on EHS matrix were also triple stained for cytokeratin 14, cytokeratin 18, and cytokeratin 19, and for cytokeratin 14, cytokeratin 18, and α-isoform smooth muscle actin. The anti-cytokeratin 19 antibody LP2K stains all mouse luminal epithelial cells in vivo but only stains lumen-derived clones heterogeneously in vitro (Smalley et al. 1998).
The results showed that approximately 50–90% of the cytokeratin 18-positive cells in Type I and Type II colonies on EHS were weakly or moderately stained for cytokeratin 19 (not shown). In mammospheres, all cytokeratin 18-positive cells were strongly cytokeratin 19-positive, which again confirms their specific luminal identity (not shown). Lumen-derived clones showed little or no staining for α-smooth muscle actin (not shown).
Comparable results were obtained from nine separate 33A10 sorts from different batches of mice, each of which was simultaneously plated with and without EHS matrix, and from 13 separate preparations of unsorted populations of virgin mouse mammary epithelial cells. As stated above, clonal identities were established solely on the basis of sorted cells, and unsorted clones were used merely to increase the numbers of analyzable colonies. Unsorted populations were not used in the definition of clonal identities. Furthermore, all clone types that were observed in the sorted lumen-derived cultures were seen in the unsorted populations, and there were no extra colony types seen in unsorted populations that were not observed in either the sorted lumen-derived or myoepithelial populations.
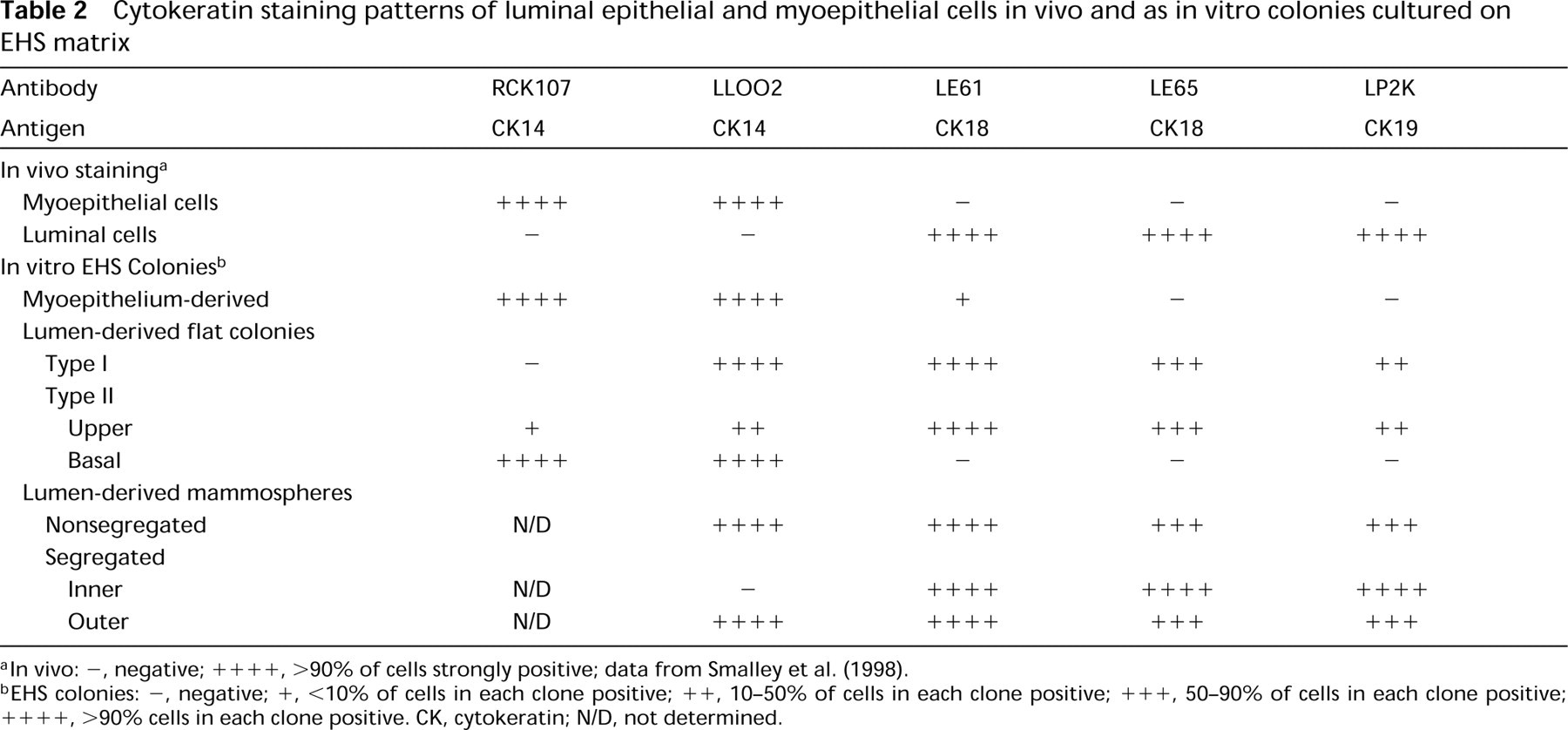
The cytokeratin staining patterns of the main clone types observed are summarized in Table 2 and shown schematically in Figure 6.
Functional Differentiation of Lumen-derived Colonies
The above results show that the cloned luminal cells on EHS matrix developed a structure and differentiated pattern of cytoskeletal antigen expression that resembled that of intact mouse mammary parenchyma, i.e., a cell layer in Type II (multilayer) flat clones with strictly basal characteristics and inner cells in mammospheres with a strictly luminal phenotype. The capacity of these structures to respond to lactational stimuli was therefore examined by β-casein staining of clones cultured for 8 days on glass or on EHS matrix in the basic growth medium or in lactation medium (see Materials and Methods). Only clones in the cultures grown on EHS in lactation medium were stained by the antibody to this protein. Furthermore, staining was seen only in mammospheres, not in flat clones, and only in the outer layer of the spheres nearest the gel, not in the center of the mammosphere (Figure 7).
Discussion
We have used flow sorting, cell cloning, and multiple indirect immunofluorescence analysis as a novel method of investigating the role of ECM in the definition and maintenance of the differentiated cellular identity of virgin mouse mammary luminal epithelial and myoepithelial cells. It has previously been observed (Smalley et al. 1998) that such cells dedifferentiate when they are established in clonal culture on glass or tissue culture plastic and promiscuously express cytoskeletal markers that are cell type-specific in vivo. This phenomenon is not the result of genetic drift but is epigenetic because it is seen within 24 h after separated luminal epithelial cells are placed in culture and after 6 days in the case of separated myoepithelial cells. To address the question of whether the ECM reinforces cell identity in the mouse mammary gland and whether it is the lack of ECM in culture that results in deregulation of the cell phenotype, separated mouse mammary luminal epithelial and myoepithelial cells were cultured on EHS matrix. The clones that grew from these cells were characterized with regard to their morphology and cytoskeletal staining pattern, as indices of loss or maintenance of differentiated cellular identity. The results indicated a role for both cell-cell and cellmatrix interactions in maintaining specific epithelial phenotypes.
Cytokeratin staining of mouse mammary luminal epithelial three-dimensional clones on EHS matrix. Cultures were double stained with LLOO2 (cytokeratin 14; A,C,E) and LE61 (cytokeratin 18; B,D,F). (A,B) Mammosphere with nonsegregated staining. (C,D) Mammosphere showing segregated staining. Bar = 35 μm. (E,F) Confocal image of segregated mammosphere. Bar = 30 μm.
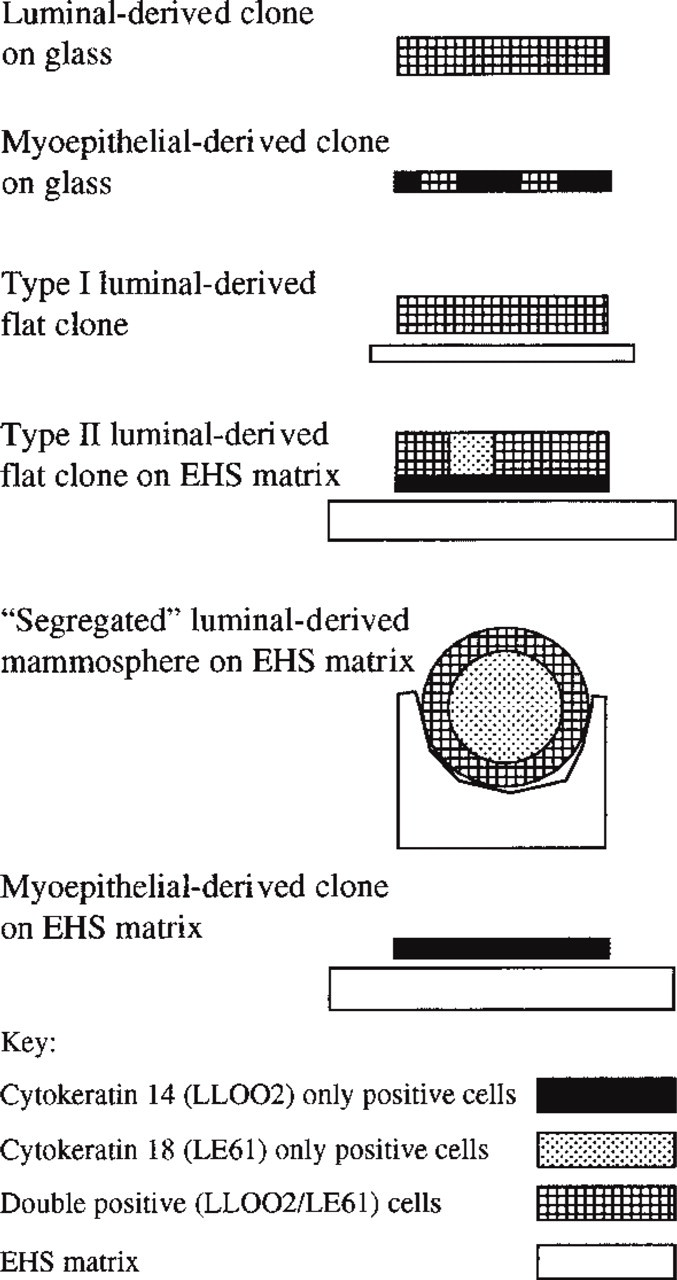
Schematic illustration of the cytokeratin 14 and 18 staining patterns of the major clone types derived from mouse mammary epithelial cells cultured on glass (data from this study and from Smalley et al. 1998) and on EHS matrix (data from this study).
Cytokeratin staining patterns of luminal epithelial and myoepithelial cells in vivo and as in vitro colonies cultured on EHS matrix
Culture of separated myoepithelial cells as clones on EHS matrix resulted in retention of the myoepithelial identity of the cells (as defined by cytokeratin 14 expression) and to a large degree prevented the expression of an in vivo marker of luminal epithelial cells (cytokeratin 18), which is seen in corresponding clones cultured on glass. There was no evidence in this study that the adult myoepithelial cells could generate a luminal cell type in vitro; structures resembling Type II clones were never seen in sorted myoepithelium cultures. In contrast to the uniformity in phenotypes seen with the myoepithelium-derived clones, those derived from purified luminal epithelial cells showed diverse morphologies and staining patterns, indicating a greater complexity in the factors that determine the specific phenotypes of cells in these clones. In particular, the Type II (multilayer) clones generated de novo a cell layer with a cytokeratin staining pattern (cytokeratin 14-positive/18-negative) that is typical of basal cells.
The basal cell layer of the Type II clones is unlikely to have been derived from myoepithelial cells contaminating the 33A10-positive luminal cell population. Any myoepithelium-derived cells in this situation can have arisen only as contaminants in the luminal sorts. Such contamination could occur only in two ways. First, the single-cell gating, which is the preliminary stage of the cell sorting, would have to be inefficient such that it allowed through doublets of cells (which must further have been composed of one luminal epithelial and one myoepithelial cell). However, preliminary experiments from when the system was being established showed that the flow cytometer sorted single-cells with a greater than 99% purity. The second possibility is that the pure single-cell populations contained contaminating single myoepithelial cells among the single luminal epithelial cells. Our previous data (Smalley et al. 1998) on sorted mouse mammary epithelial populations cloned as monolayers, however, showed that flow sorting was of high stringency and isolated > 99% pure luminal epithelial cells from a population of cells that was >99% single cells. The frequency at which the Type II clones arose (approximately 10% of flat clones) therefore makes it extremely unlikely that their origin is anything other than purely luminal epithelial. Furthermore, the basal cells in Type II clones did not stain for α-isoform smooth muscle actin, whereas sorted, myoepithelium-derived clones were positive for this marker.
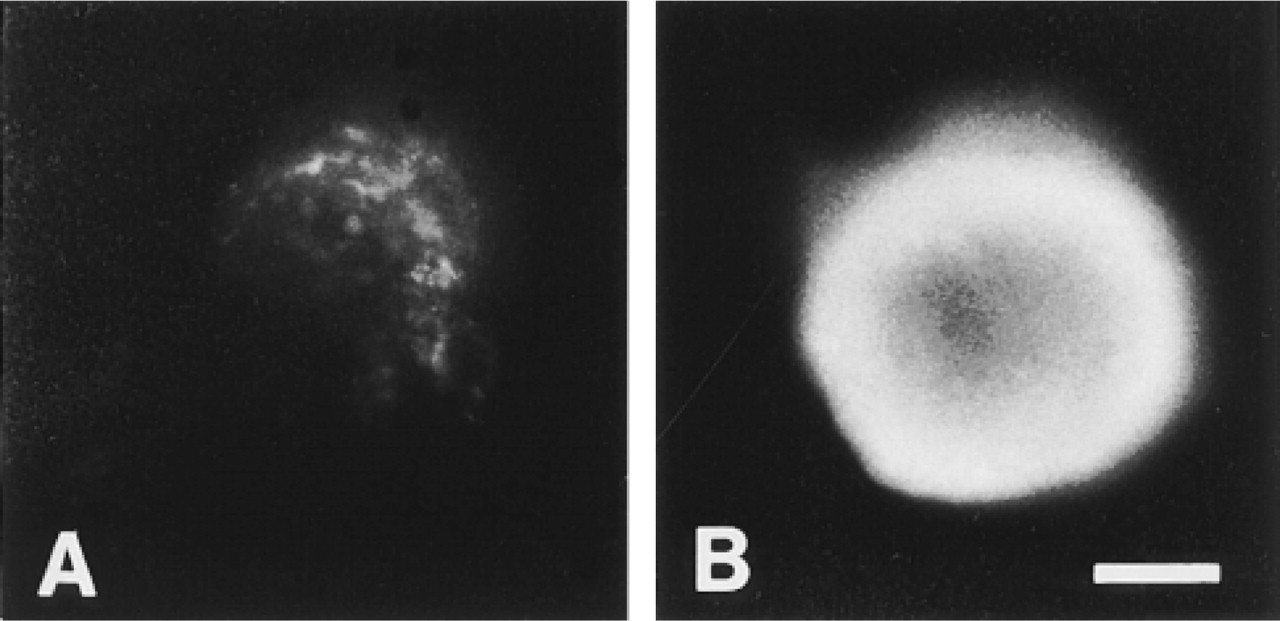
Functional differentiation of mouse mammary epithelial three-dimensional clones on EHS matrix. Mammospheres were stained with anti-casein antibody RβC-1 after culture in medium without (A) or with (B) lactogenic hormones (see text). Staining was observed only in the outer layer of the spheres that had been cultured with hormones. Bar = 65 μm.
A different pattern of segregation and differentiation of cells was seen in the mammosphere type of luminal clone. In the majority of these, an exclusively luminal phenotype (cytokeratin 18-positive/19-positive/14-negative) was seen in the central mass of cells. The outer layer of the mammospheres, however, retained the promiscuous cytokeratin expression and still stained with both luminal and basal markers (i.e., they were cytokeratin 18-positive/19-positive/14-positive), in contrast to the basal layer of the Type II (multilayer) flat clones. The cytokeratin 14/18 staining patterns of the major clone types are summarized schematically in Figure 6. It is clear that sorted single luminal epithelial cells from the virgin mouse mammary parenchyme can generate both luminal and basal epithelial cells, depending on the manner in which they interact with each other and the surrounding matrix. Luminal epithelium-derived clones could not generate fully differentiated myoepithelial cells with both basal cytokeratins and α-isoform smooth muscle actin, however, at least under the culture conditions used. For this study we limited the culture conditions to a single set of parameters that we had previously shown to allow maximal clonal growth of mouse mammary epithelium (Smalley et al. 1998), but it is entirely possible that the differentiative potential of the clones might be medium-dependent. In general terms, the effects of different medium components on the different mouse cell types have yet to be established.
We propose that the difference between the cytokeratin 18-positive/14-negative phenotype of the innermost cells in the mammospheres and the principally double-positive cells of the flat clones shows that homotypic cell-cell interactions reinforce and maintain the luminal epithelial phenotype. It may be that the topography or extent of these interactions is important in the maintenance of the luminal epithelial phenotype in this system. It is unlikely that the EHS matrix takes an active role (e.g., via ligand-receptor interactions) in the maintenance of the luminal cell identity as the central mass of cells in the mammospheres, which exclusively expressed the luminal cell markers, were not in contact with it. However, it does appear that the extracellular matrix has an active role in promoting the generation of de novo basal epithelial cells (in the Type II flat clones).
At first sight it appears paradoxical that the outermost cells of the mammospheres are apparently less well differentiated as basal cells than their counterparts in the Type II (multilayer) flat clones. However, as shown by SEM, most of the basal cell layer of the mammospheres was covered only by a very thin matrix layer and in some areas was not covered by matrix at all. The Type II (multilayer) flat clones, in contrast, were always observed on a well-defined matrix layer, under which conditions a cytokeratin 14-positive/18-negative basal layer formed, and it is therefore likely that active ECM signaling played a role in the generation of this layer.
This study provides evidence, therefore, that mouse mammary luminal epithelial cells are, or contain, a compartment that is pluripotent, as shown by their ability to generate clones that possess a basal cell layer lacking luminal cytoskeletal markers, although it does not demonstrate conversion from luminal epithelial cells to fully differentiated myoepithelial cells. These findings support the work of Pechoux et al. (1999), which showed that a subset of mature human breast luminal epithelial cells gave rise to myoepithelial cells over an extended culture period. It is impossible to say whether all lumen-derived cells could generate Type II clones if they were given the appropriate, ideal conditions. We therefore cannot state whether the ability to generate a basal cell layer de novo is a property shared by all mouse luminal epithelial cells or whether it is found in a subpopulation of these cells. Several immunocytological (e.g., Sonnenberg et al. 1986; Christov et al. 1993; Zeps et al. 1998) and ultrastructural (Russo et al. 1976; Chepko and Smith 1997) studies of rodent mammary epithelium have implied that there are subpopulations within the luminal epithelium, as does the work of Pechoux et al. (1999). They support the view that the Type II progenitor cell is a subpopulation of the 33A10-positive luminal epithelium.
The above model, in which cell-cell interactions reinforce luminal cell identity and contact with an optimal amount of ECM is required to generate basal epithelial cells de novo from luminal epithelial cells, must also accommodate the observation that culture of mammospheres in a lactation medium resulted in β-casein production only in the outermost cell layer, in which we have demonstrated stains for both the luminal epithelial and basal markers, and not in the more fully luminally differentiated central cells of the mammospheres. It is known that mouse mammary luminal epithelial cells isolated in vitro require contact with ECM for functional differentiation (Streuli 1993) and that the functional unit of lactation in vivo, the alveolus, is not totally covered with myoepithelial cells. Instead, these form a basket-like network (Emerman and Vogl 1986) through which the underlying secretory luminal cells can contact the matrix. It is therefore likely that β-casein expression occurs in the outer cells of mammospheres because only these cells are in contact with a (thin) layer of ECM. Because they still have both luminal and basal characteristics, they are still competent to respond to hormonal stimuli of lactation, although we cannot definitively exclude the possibility that there may be problems with diffusion of lactogenic hormones into the interior of mammospheres. The fact that β-casein production occurred only in mammospheres after 8 days but not in Type II flat clones, the cells of which possess both luminal and basal characteristics and which are in contact with matrix, indicates that there are further influences over functional differentiation, possibly including the increased cell-cell contact that occurs even on the outer layer of spheres. The interaction of three-dimensional morphology and functional differentiation has been demonstrated previously by Chen and Bissell (1989), Smalley et al. (in press), and Close et al. (1997). If such cell-cell contact does have a role in functional differentiation, it is perhaps in the initiation of the process or in priming of cells to be able to respond to lactogenic signals rather than in the maintenance of lactation, because Streuli et al. (1991) have demonstrated that single cells derived from trypsinization of monolayer cultures of mammary epithelial cells harvested from midpregnant mice can produce β-casein in the presence of lactogenic hormones when embedded in EHS matrix.
We have demonstrated in this study the utility of single cell sorting and clone-by-clone analysis of cell type-specific markers in the dissection of the differentiative capacity of the component cells of a complex tissue, and we have provided in vitro evidence from primary mouse mammary epithelial cells for the existence of pluripotential cells within the mammary luminal epithelial but not the myoepithelial cell compartment. Our study supports the work of Pechoux et al. (1999) and complements recent in vivo experiments using a serial mammary epithelial transplant model which demonstrated that an entire functional mammary gland could be derived from the progeny of putative single multipotent stem cells (Smith 1996; Kordon and Smith 1998). Furthermore, the results we have presented argue for a cell type-dependent variability in the importance of cell-matrix and cell-cell interactions on cell differentiation in the context of organogenesis.
Footnotes
Acknowledgements
Supported by the Cancer Research Campaign. We would like to thank Professor Barry Gusterson for his support.