
Abstract
Small leucine rich proteoglycans (SLRPs), including Biglycan, have key roles in many organ and tissue systems. The goal of this article is to review the function of Biglycan and other related SLRPs in mineralizing tissues of the skeleton. The review is divided into sections that include Biglycan’s role in structural biology, signaling, craniofacial and long bone homeostasis, remodeled skeletal tissues, and in human genetics. While many cell types in the skeleton are now known to be affected by Biglycan, there are still unanswered questions about its mechanism of action(s).
Introduction
Small leucine rich proteoglycans (SLRPs), including Biglycan, have key roles in many organ and tissue systems. The goal of this article is to review the function of Biglycan and other related SLRPs in mineralizing tissues of the skeleton. The review is divided into sections that include Biglycan’s role in structural biology, signaling, craniofacial and long bone homeostasis, remodeled skeletal tissues, and in human genetics. While many cell types in the skeleton are now known to be affected by Biglycan, there are still unanswered questions about its mechanism of action(s).
Bone Biology
The skeletal system consists of cartilage, joints, bones and associated soft tissues, such as tendons, ligaments and marrow. Bone is a unique tissue composed of several cell types associated with a 3-dimensional mineralized extracellular matrix (ECM). It differs from other tissues in the body in that it is largely mineral and because a uniquely large proportion (approximately 90%) of its total organic material is collagen (mainly type I). Of the non-collagenous proteins, a large proportion are proteoglycans (PGs), responsible for 10%–15% of the ECM’s wet weight (where water and dissolved matrix molecules represents 65%–80% of the total weight and collagen constitutes about 10%–20% of the ECM wet weight)1–3
The skeleton is a dynamic tissue undergoing continuous regeneration known as bone remodeling, in which bone is being resorbed and formed simultaneously at many discrete foci. This renewal process has a key role in the maintenance of the skeleton and in its physiologic functions, such as body growth, hematopoiesis, adaptation to changing mechanical loads, renewal of the mineralized matrix, and the regulation of the body’s calcium-phosphate reservoir.4,5
The cellular component of bone arises from two distinct lineages. The osteogenic lineage gives rise to the stroma, bone-forming cells, osteoblasts and their descendants, lining cells and osteocytes, whereas the hematopoietic lineage gives rise to bone resorbing cells, osteoclasts.
Osteoblasts are responsible for the synthesis of the bone ECM (osteoid) and its subsequent mineralization. As they mature, these cells begin to produce bone-enriched proteins, growth factors and hormones.
Osteoclasts are short lived, mobile, multinucleated giant cells derived from the fusion of monocytic lineage cells. Osteoclasts are usually in direct contact with a calcified surface (hypertrophic cartilage and bone) and within a resorption bay (Howship’s lacunae), as a result of their own resorptive activity. Osteoclasts actively resorb bone by creating a sealed zone (an “extracellular lysosome”) where acidification and chelation of calcium occurs, thus destroying the hydroxyapatite crystals, which leads to unmasking of ECM proteins and their processing and/or degradation by lysosomal enzymes such as tartrate-resistant acid phosphatase (TRAP) and cathepsin K (CTSK). The calcium, phosphorous, and matrix degradation products are transferred by the osteoclast to the blood circulation. 6
Skeletal Development and Bone Turnover
Embryonic skeletal development arises from two distinct developmental processes: (1) intramembranous ossification, in which bone is generated directly from osteoblastic cells without an intermediate cartilage template and (2) endochondral ossification, which involves synthesis of an intermediate cartilaginous template. Postnatally, bone undergoes constant remodeling, whereby the osseous mineralized ECM is continuously being replaced simultaneously at many discrete focal points. A balance must exist between the overall volume of bone resorbed and formed during remodeling in order to maintain optimized bone structure and strength and to ensure correct mineral homeostasis. 4
Both (embryonic) skeletogenesis and remodeling are complicated processes spatially and temporally regulated by numerous central, systemic (endocrine), and local factors. Alterations of these regulatory mechanisms can disturb the balance between resorption and formation and lead to metabolic bone diseases, such as osteoporosis.7,8
Proteoglycan Structure and Growth Factor Modulation
PGs are macromolecules composed of a protein core to which at least one glycosaminoglycan (GAG) side chain is covalently bound to a serine residue. GAG chains are linear, negatively charged polysaccharides. Based on the presence of hexosamine isomers, GAG chains can be divided into two distinct groups: glucosaminoglycans which have
Accordingly, it is critical to understand the role PGs play in (musculo)skeletal biology, particularly SLRPs, the most abundant type of PGs in bone.5,27 Therefore, we focus here on the role of SLRPs, especially Biglycan, in skeletal development and maintenance.
SLRPs: A Family With a Role in Collagen Fibrillogenesis
The SLRP family is composed of 19 members that have been classified into five distinct classes according to their amino-acid structural sequence, functional properties and chromosomal organization (Table 1). The five classes are divided into canonical (Classes I–III, containing 14 members) and non-canonical classes (Classes IV–V, including 5 members).27,28 The tandem leucine-rich repeat (LRR) motif in the core protein of SLRPs is particularly important for binding different types of collagen, both fibrillar and non-fibrillar.19,27,29–36 In fact, different stages of fibril formation require different SLRPs.13,30,37–40 The GAG moieties also play an important role in collagen fibrillogenesis and preservation.13,14
Organizational Class System of SLRP Family Members.
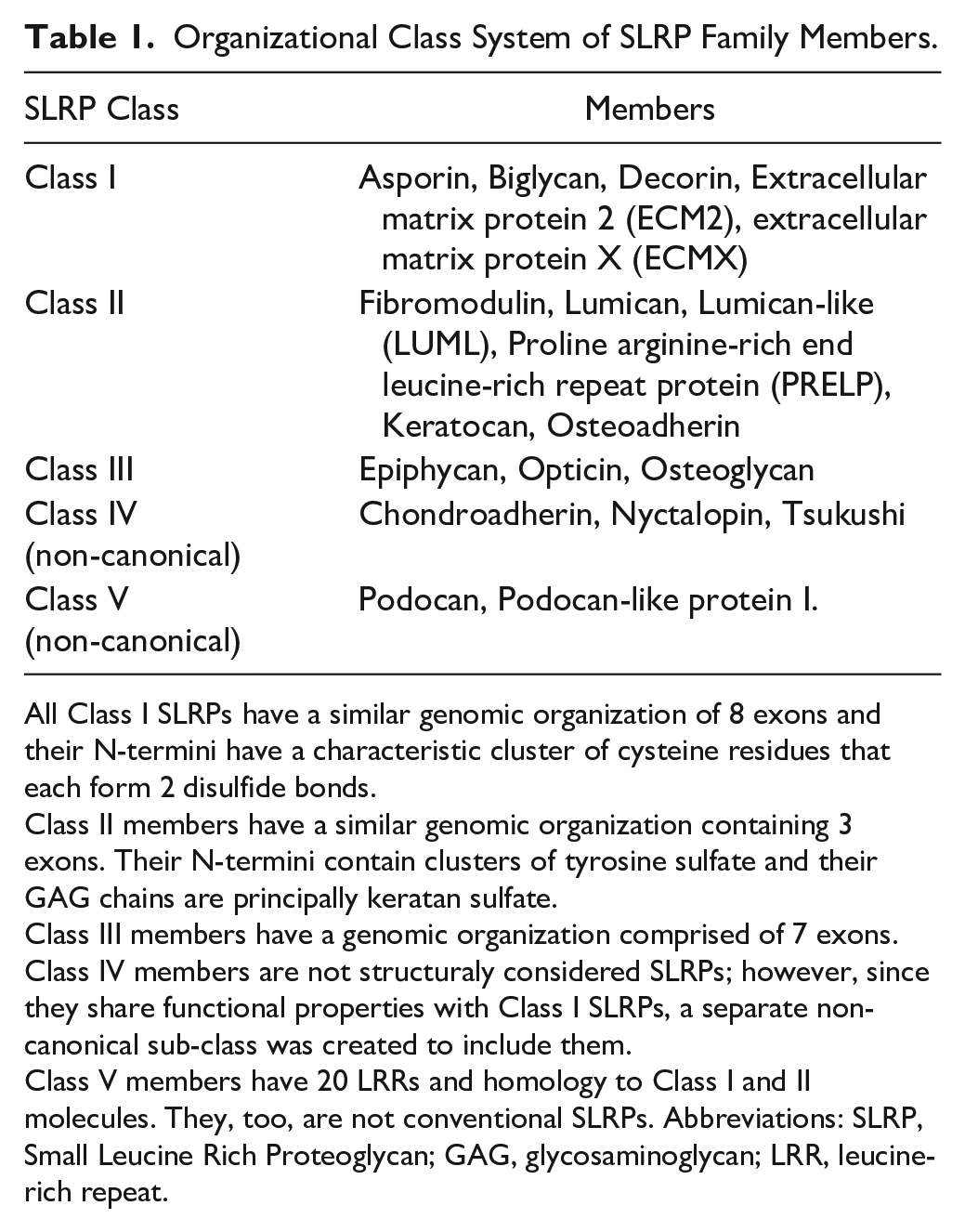
All Class I SLRPs have a similar genomic organization of 8 exons and their N-termini have a characteristic cluster of cysteine residues that each form 2 disulfide bonds.
Class II members have a similar genomic organization containing 3 exons. Their N-termini contain clusters of tyrosine sulfate and their GAG chains are principally keratan sulfate.
Class III members have a genomic organization comprised of 7 exons.
Class IV members are not structuraly considered SLRPs; however, since they share functional properties with Class I SLRPs, a separate non-canonical sub-class was created to include them.
Class V members have 20 LRRs and homology to Class I and II molecules. They, too, are not conventional SLRPs. Abbreviations: SLRP, Small Leucine Rich Proteoglycan; GAG, glycosaminoglycan; LRR, leucine-rich repeat.
The continued presence of a SLRP “coating” on collagen fibrils, even after the collagen fibers are assembled, prevents overgrowth of the fiber beyond a certain diameter and protects against collagenase activity, thus enabling proper matrix function.30,41 For example, it was recently demonstrated that the interaction between the GAG chains of SLRPs and collagen fibrils under acidic conditions (like those found in an osteoclast resorption pit microenvironment), helps retain the fibril structure and regulates cathepsin K-mediated bone degradation. 42
Functional Implications of Post-translational Modifications
Post-translational modifications, such as glycanation, are also essential in the early stages of collagen fibrillogenesis. For instance, the core protein of Biglycan does not bind to collagen as strongly as a related Class I SLRP, Decorin37,38,43,44; however, the PG version of Biglycan (core protein with two GAG chains) binds much more strongly to collagen. 13 Fig. 1 portrays the role of Biglycan in collagen fibrillogenesis. Moreover, lack of Decorin and/or Biglycan glycanation severely impairs collagen fibrillogenesis.45–47 Sulfation of the tyrosine residues is another type of posttranslational modification that affects SLRPs’ interactions with collagen and non-collagenous proteins. Fibromodulin, a Class II SLRP, can bind to collagen via its LRR and a sulfated tyrosine domain at the N-terminus, allowing it to simultaneously interact with two collagen molecules, affecting collagen fibril formation as well as arrangement of collagen molecules into highly organized fibrillar structures.48–50 Additionally, Fibromodulin’s affinity for heparin-binding proteins is dependent on the number and position of the sulfated tyrosine residues. 50 Since HS is a functional component of the RANKL/RANK/OPG axis, involved in osteoblast-osteoclast crosstalk, Fibromodulin may be involved in this axis through its sulfated motifs.
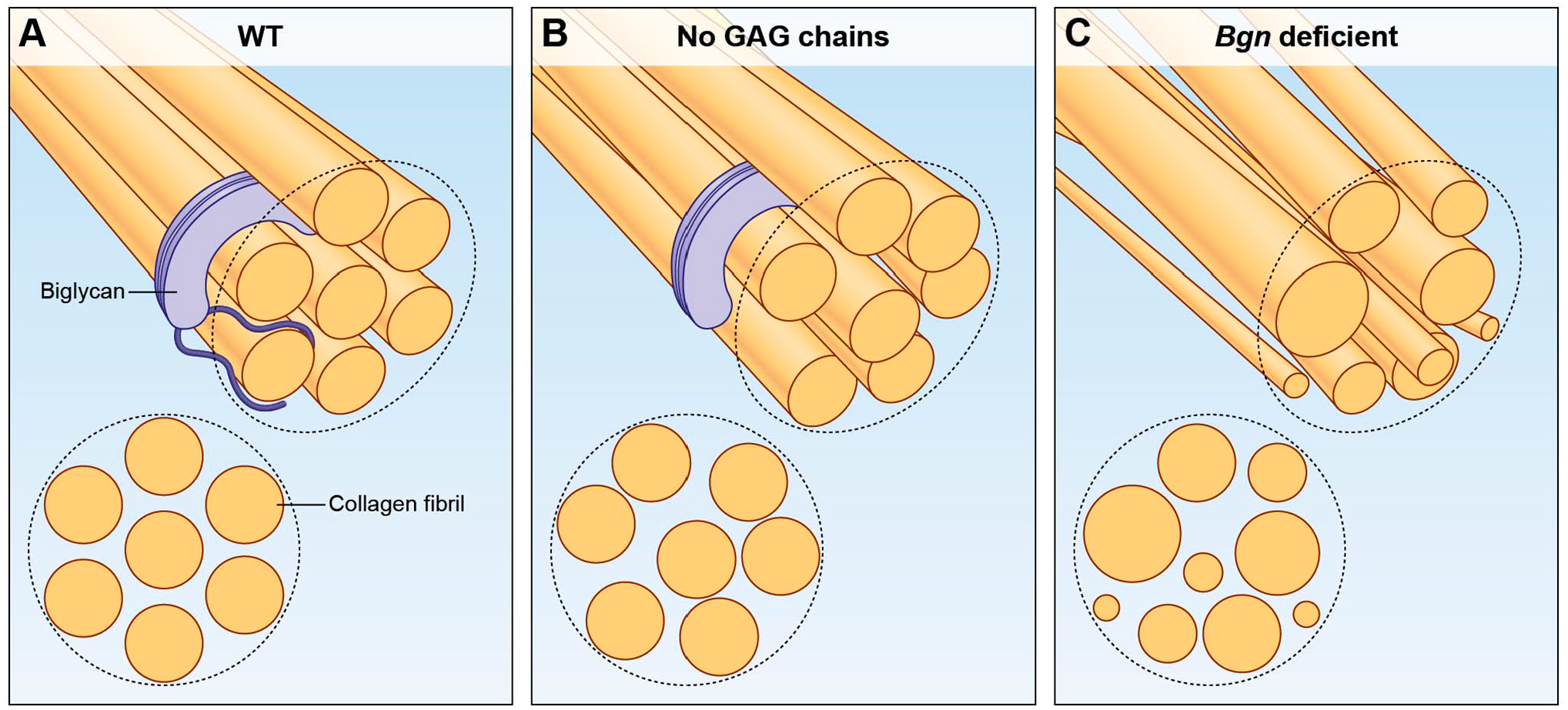
SLRPs, such as Biglycan, bind collagen fibrils, either through the core protein or the GAG chains thereby regulating the diameter of the individual fibrils as well as their spatial assembly into thicker bundles. (A) Depicts a collagen fiber, and cross section, when Biglycan is present in its glycosylated form. (B) Post translational modifications to the glycanation state of SLRPs, e.g. total loss of GAG chains, results in structural abnormalities of the collagen fiber. (C) Lack of even a single type of SLRP, impairs collagen fibrillogenesis as well as arrangement of collagen molecules into highly organized fibers. Abbreviations: GAG, glycosaminoglycan; SLRPs, small leucine rich proteoglycans.
SLRP Expression and Localization During Development and Aging
During skeletal development and establishment of postnatal homeostasis, extensive variations in the types and amounts of extracellular macromolecules that are synthesized and expressed are seen. Different spatial patterns of expression of Decorin and Biglycan are found in mature and immature bones, pointing to the different roles these two SLRPs play at specific sites during distinct developmental events, such as initial osteoid formation and subsequent bone mineralization. During skeletal development, high levels of Decorin and Biglycan mRNA are detected in areas of endochondral and membranous bone formation.51,52 Decorin is mainly seen in the central areas of resting cartilage of the diaphysis, whereas Biglycan expression is limited to osteoblasts and a rim of chondrocytes close to the articular surface. In immature bones, Decorin is expressed throughout the osteoid matrix and is associated with osteogenic and non-osteogenic layers of the periosteum, whereas in mature bone, Decorin is present in the perilacunar matrix, the canaliculi of osteocytes, and the matrix immediately adjacent to quiescent Haversian canals (which are a network of tubes in the cortical bone allowing the passage of blood vessels and nerves). Biglycan, on the other hand, is localized to the walls of osteocyte lacunae (where it may also act as a transducer of sheer forces) and bone cell surfaces within developing bone, but appears to be evenly distributed throughout the bone matrix in mature bone.51–53 Fibromodulin is expressed by both chondrocytes and osteoblasts during fetal endochondral and intramembranous bone development. In the growth plate, Fibromodulin appears together with Biglycan in the reserve and proliferative zones54,55 and Fibromodulin deposition is also found around the late-hypertrophic chondrocytes of the secondary ossification center.56,57
With aging, changes in PG secretion and post-translational modifications influence their binding to collagen. In fact, the glycanation state of SLRPs can serve as a chronological age “footprint.”58–61 In mineralized matrix, the synthesis of Biglycan is reduced with age, and though the abundance of the PG form of Biglycan in bones appears to be stable throughout life, in articular cartilage and the intervertebral disc, there are noticeable changes: the GAG-free form of Biglycan is low in juveniles, but increases in adults until it becomes the predominant form.62,63 For Decorin, though the GAG-free form is always a minor component, the level of glycosylated Decorin decreases with age, thereby slightly increasing the proportion of the GAG-free form.63,64 Like Decorin and Biglycan, the GAG chains of Fibromodulin are shortened with aging until the GAG-free form becomes the predominant one in mature cartilage. 65 These changes can be the result of more rapid degradation of the glycanated precursors or from changes in growth factor/cytokine synthesis with aging. 66 The composition of the GAG chains attached to the SLRP core protein also changes during bone development. Both Decorin and Biglycan change from dermatan to CS chains as the osteoblasts move along the differentiation path from the cell proliferation phase to the mineralization phase. 67
Post-translational Modifications and Pathology
Despite evidence that SLRPs without GAGs are present in physiological settings (as was mentioned before), modifications in the glycanation states of SLRPs may also correlate with pathologies. For instance, in gerodermia osteodysplastica (GO; OMIM: # 231070), characterized by early onset osteoporosis among other phenotypes, there is defective glycanation of Decorin and Biglycan. 64 Another example is degenerative suspensory ligament desmitis (DSLD), which resembles Ehlers–Danlos syndrome (EDS). In this disease, the accumulation of Decorin carrying abnormal GAG chains leads to reduced TGF-β1 binding affinity, high levels of bone morphogenetic protein 2 (BMP-2) and the production of antibodies against Decorin.68–70
SLRPs and Signal Transduction
In addition to being structural proteins, SLRPs constitute a network for signal regulation. They control cell proliferation and differentiation in a cell-specific fashion and modulate distinct pathways via intracellular phosphorylation, a major conduit of information for cellular responses.27,71–73 For example, low Biglycan levels decrease osteoblast maturation accompanied by an increase in proliferation of osteoblasts for compensatory purposes. This, in turn, leads to a decreased amount of osteoblast progenitors.29,74–76 Additionally, both Biglycan and Fibromodulin regulate osteoclast differentiation through their effect on osteoblasts and their precursors. Though absence of Biglycan lead to increased serum levels of both OPG and RANKL, which regulate osteoclastogenesis, the end result is increased osteoclast differentiation and osteolysis.57,77 Biglycan also interacts with osteoclast precursors (macrophages) and conveys inflammatory processes through pathogen recognizing receptors.22,78
SLRPs are also known to be involved in TGFβ/BMP activity, receptor tyrosine kinases (such as ErbB family members), insulin-like growth factor I receptor, and Toll-like receptors signaling cascades.15–18,22,71,79 Both Decorin and Biglycan bind all 3 isoforms of TGF-β. Bone cell extract analysis revealed that TGF-β is mainly bound to Decorin’s core protein and that the interaction between TGF-β and Decorin actually increases the affinity of TGF-β to its receptors and as a consequence, its activity. Lack of Biglycan resulted in a reduced response to TGF-β by bone marrow stromal cells.15,17,80–82 Interestingly, TGF-β members have a positive feedback effect, as all 3 isoforms of TGF-β were shown to upregulate the expression of both Biglycan mRNA and protein. 83
Biglycan also directly binds members of the BMP family. The core protein of Biglycan enhances BMP-2 activity, whereas the GAG chains enhance the binding of BMP-4 to osteoblasts. Consequently, Smad 1/5/8 phosphorylation is increased resulting in the expression of osteoblastic markers such as Runx2, Osterix (OSX), Osteopontin (OPN/SPP1), Bone Sialoprotein (BSP), and Osteocalcin (BGLAP) and ultimately osteogenesis.17,18,84,85 Biglycan also plays a crucial role in activating the MAPK pathways (ERK and p38 signaling pathways) promoting osteoblast differentiation. This activation is mediated through the GAG chains, as phosphorylation of Erk is not observed when the GAG-free form of Biglycan is presented to pre-osteoblasts or macrophages.23,78 Furthermore, Biglycan was shown to trigger the Wnt pathway by binding to both Wnt3a as well as the Wnt receptor, LRP6/5. Though the core is more effective, both glycosylated and non-glycosylated forms can activate this signaling pathway.86,87 As described above, aging affects the GAG chains’ structure and post-translational modifications, and such changes consequently affect the different signaling pathways and alter differentiation, proliferation, and maintenance of bone.
Animal Models
Skeletal manifestations are observed in mouse models in which specific SLRP genes were ablated or altered (leading to altered post-translational modifications), in some cases paralleling clinical phenotypes of human diseases, that is, in spite of compensatory expression of other members of the same sub-class of the ablated SLRP gene.44,88,89 Many of the clinical phenotypes are associated with abnormal collagen fibril size, shape, and cross-linking.13,30,37–40 Though Decorin is expressed in bone, Decorin-deficient mice did not manifest any changes to bone mass or architecture.76,90,91 Biglycan-deficient mice fail to achieve peak bone mass and develop age-dependent osteopenia. Phenotypic analysis of these mice revealed that the Bgn-KO mice have decreased numbers of bone marrow stromal cells (a subset of which are skeletal stem cells), leading to lower osteoblast number and activity, increased rate of osteoclast differentiation and activity, reduced response to TGF-β, BMP-2, and BMP-4 signaling; reduced collagen synthesis; and relatively more apoptosis compared with cells derived from normal WT mice.17,18,74,76,77,92
Mutual Compensation of SLRPs
Due to the structural similarity of Decorin and Biglycan, a compensatory mechanism is triggered when either is ablated, leading to an increased expression of the other. This overexpression of other SLRP members could, at least to some extent, be explained by clustering of SLRP family members genes on their respective chromosomes. When double knockout mice (Dcn/Bgn-DKO) were generated, it led to a more severe skeletal phenotype than the single KO models. The skeletal manifestations in these Dcn/Bgn-DKO mice developed earlier and affected both the cortical and trabecular bone mass as well as cranial suture fusion.76,92,93 This phenotype resembles the clinical manifestations of the progeroid form of EDS (OMIM:#130070),45,47,76,92 a group of heritable connective tissue disorders that share the common features of skin hyperextensibility, articular hypermobility, and tissue fragility. The molecular mechanism appears to be based on increased TGF-β signaling, which shifts a proportion of bone marrow stromal cells from growth to apoptosis, leading to fewer osteoprogenitor cells and subsequently reduced bone formation.76,92,94
Fibromodulin-ablated (Fmod-KO) mice demonstrate significant ectopic ossification, higher bone mass, and early-onset osteoarthritis phenotype.55,95 These mice express high levels of Lumican, also a Class II SLRP, demonstrating again a compensatory mechanism.96,97 Even though differentiating and mature osteoblasts express and secrete Lumican, Lumican-deficient (Lum-KO) mice do not exhibit a bone phenotype.98,99 Fmod/Lum double null mice demonstrate gait abnormalities, joint laxity, misaligned patellae, severe knee dysmorphogenesis, and extreme tendon weakness, all leading to age-dependent osteoarthritis,92,97,99 phenocopying the clinical manifestations of EDS Spondylodysplastic Type 1 (OMIM #130070).
Bgn/Fmod double-KO mice have markedly low bone mass (LBM), which is a result of high levels of TNFα and RANKL being expressed and secreted by their osteoblasts. Due to the lack of these two SLRPs, both cytokines are not sequestered in the cellular microenvironment and are “free” to instigate rampant osteoclastogenesis. 57 Fig. 2 illustrates the effect that TNF-α overactivity has, due to lack of Biglycan, on both osteoblasts and osteoclasts.
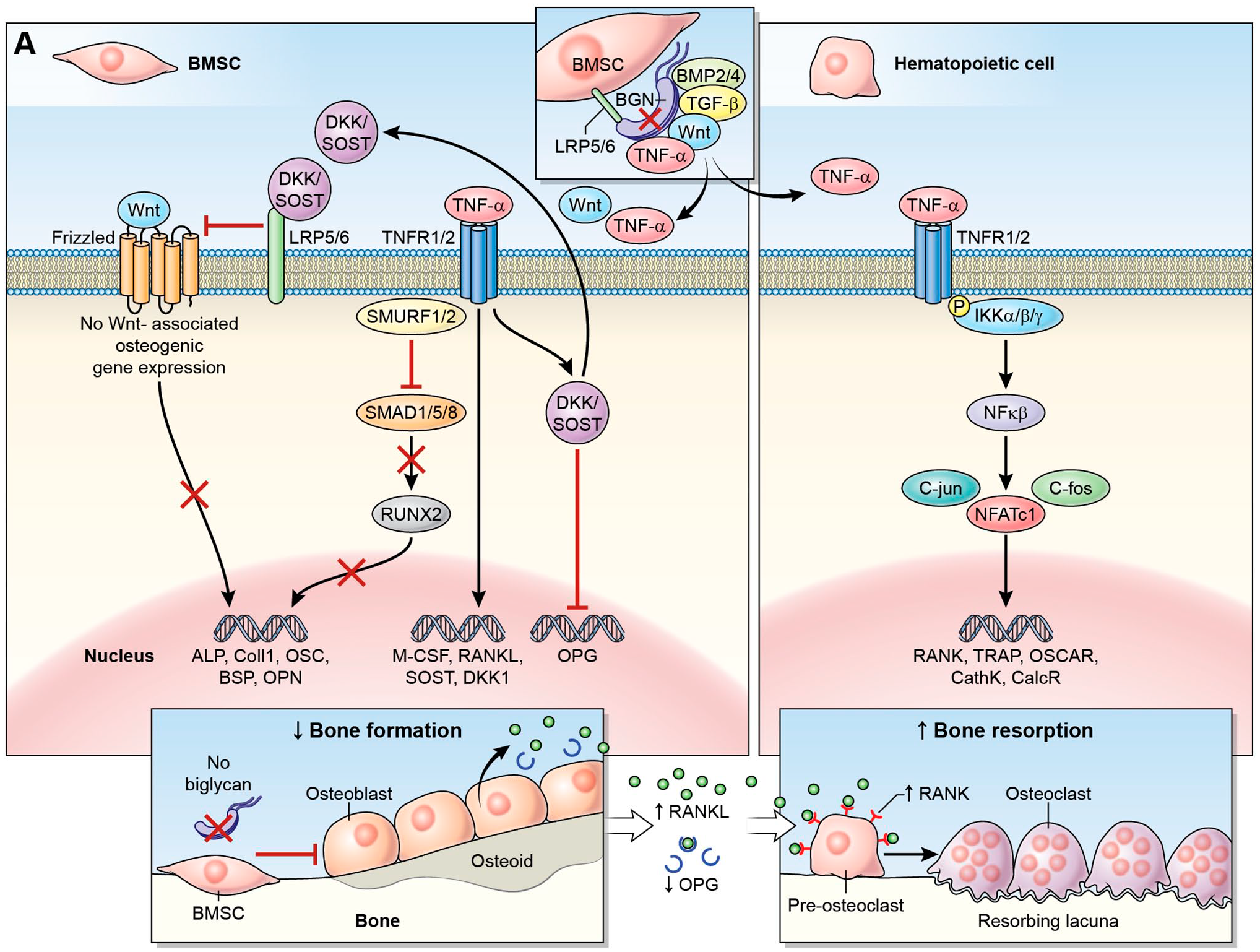
Bone marrow stromal cells express and secrete Biglycan, which directly binds many growth factors, morphogens, cell surface receptors and other ECM structural and adhesive molecules. When Bgn (or any other SLRP) is missing, these cytokines are not being sequestered in the cellular microenvironment, thus modifying the closely regulated signal transduction of both osteoblasts and osteoclasts. Here, we focus on the effect of TNFα overactivation. TNFα binds to its receptor on the membrane of both BMSCs (A) or hematopoietic/preosteoclastic cells (B). A. In pre-osteoblasts, TNFα activates Smurf1/2, inhibiting SMAD1/5/8 phosphorylation, which leads to the inhibition of osteogenic gene expression regulated by the osteoblastogenesis transcription factor, RUNX2. TNF-α also induces the expression of Dickkopf-1 (DKK-1), an inhibitor of Wnt signaling, leading to a decrease in bone formation. Furthermore, Canonical wnt signaling promotes OPG expression, thus the inhibition of this pathway leads to decreased OPG levels. TNF-α activation directly increases the expression M-CSF and RANKL by BMSCs. B. in pre-osteoclasts, TNF-α signaling leads to phosphorylation of the IKK complex with subsequent phosphorylation and proteolytic breakdown of IκB, an inhibitor of the NFκB. The activated NFκB, together with C-fos, c-jun, increase the experssion and activity of NFATc1, the master transcription factor in osteoclast differentiation, which then translocates into the nucleus, increasing the expression of osteoclast target genes such as TRAP, calcitonin receptor, cathepsin K and OSCAR. TNF-α signaling also enhances preosteoclast expression of RANK and c-fms, the M-CSF receptor. The combined effect is a decrease in bone formation signals with an increase in osteoclast differentiation, activity and survival, resulting in a low bone mass phenotype. Abbreviations: BGN, Biglycan; BMSC, bone marrow stromal cells; BSP, bone sialoprotein; ECM, extracellular matrix; OPN, osteopontin; SLRP, small leucine rich proteoglycan; TRAP, tartrate-resistant acid phosphatase.
The “Challenged” Skeletal System
Specific life events put the skeletal remodeling process under stress and challenges the ability of both osteoclasts and osteoblasts to react and restore homeostasis. Different experimental models have been devised during the years to mimic these situations. For example, increased mechanical loading such as intense exercise is known to enhance bone mass, decrease bone fragility and improve the quality of the ECM. 100 Therefore, it was suggested by Wallace et al., to improve the poor bone quality of Bgn-KO mice by subjecting them to an exercise regimen. Because genetic background is a crucial factor in the response to mechanical loading (e.g., bones from WT mice on a Bl6;129 background were more responsive to running than WT mice on a C3H background), Bgn-KO mice on these two genetic backgrounds were used. The total body mass of Bl6;129 Bgn-KO mice was not altered after exercise compared with Bl6;129 WT; however, exercise significantly increased the yield stress and carbonate/phosphate ratio and decreased the crystallinity of Bl6;129 Bgn-KO bones compared with Bl6;129 WT mice, thus increasing the resistance of Bl6;129 Bgn-KO bones to fracture. No differences were found between C3H Bgn-KO and their WT counterparts. 101 Taken together this suggests that the contribution of “nature” (genetic background) outweighs the “nurture” effect (exercise), at least in this scenario.
Gonadal hormones (sex hormones including estrogens, androgens and progesterone) also play a vital role in bone metabolism.102–105 In humans and mice, the decline in ovarian secreted estrogen during menopause or as a consequence of surgical intervention (ovariectomy; OVX), result in accelerated BMD reduction and enhanced fracture risk. Surprisingly, when Bgn-KO mice underwent OVX they did not show increased bone turnover or increased bone loss at 4 weeks post-surgery, whereas the WT controls lost bone mass. Moreover, urinary deoxypyridinoline (dPYR), a bone resorption marker known to be elevated after estrogen depletion, was increased in WT OVX mice compared with WT shame mice. However, no differences in dPYR levels were found between Bgn-KO shame and Bgn-KO OVX mice. Taken together, it appears that the Bgn-KO mice are protected from bone loss resulting from estrogen depletion. 106
Fracture healing is the ultimate challenge to bone remodeling. The healing process is a sequence of physiological events, including hematoma development, inflammation, callus formation, neovascularization, and osteoclastic and osteoblastic activation, culminating in the remodeling and restoration of the broken bone. 107 Biglycan has been shown to have a role in the healing process, as its expression is highly elevated in the healing callus 14 days post-fracture. Compared with WT mice, Bgn-KO mice have a smaller callus 14 days post-fracture with less cartilage tissue, indicated by reduced expression of both Aggrecan and Type I collagen. In addition, when RNA was extracted from the callus 7 days post-fracture, Bgn-KO showed lower levels of VEGF A gene expression compared with WT. 108 µCT angiography 7 days after fracture demonstrated decreased vascularity in Bgn-KO mice coupled with reduced vessel number, volume, and thickness. At this time point, endostatin, a strong angiogenesis inhibitor, was found to be colocalized with Biglycan around the new ossification site. Interestingly, 14 days post fracture endostatin expression was increased around the callus in Bgn-KO mice compared with WT mice. The in vitro endothelial cell tube formation assay demonstrated the ability of Biglycan to reverse the inhibitory effect of endostatin, which may indicate a potential role for Biglycan in angiogenesis regulation after fracture by opposing endostatin’s effect. 109
SLRPs in the Craniofacial Complex
The craniofacial skeleton develops via intramembranous ossification. In mice, Biglycan was reported to be expressed in both osteoblasts and non-mineralized areas during late stage of mandible development. 110 In new-born mice, Bgn is expressed throughout the cells of the developing tooth germ, including presecretory odontoblasts, ameloblasts, dental papilla, and stellate reticulum.111–114 Biglycan is a major constituent of unmineralized dentin (predentin), where it restricts collagen fibrillogenesis and promotes dentin formation and mineralization.115–117 In dental tissues of 5-day to 2-month old rats, the expression of Biglycan remains stable in predentin and in the pulp space, 118 whereas the expression of other SLRPs (Decorin, Fibromodulin and Lumican) have only been identified during dentin and cementum mineralization. 119 Deletion of either Bgn and/or Dcn in mice leads to hypomineralized dentin formation. 116 Bgn deletion led to a three- to fivefold thicker enamel in newborn mice; however, Bgn overexpression had no significant effect on amelogenesis. 120 Depletion of Dcn delayed enamel formation in newborn mice. Despite the strong effects induced by these deficiencies in newborn mice, there was no significant difference in enamel in mature mice. The observed “self-repair” suggests that the effects of these two SLRPs on amelogenesis are transient.116,121 The morphological differences in the enamel and dentin layers in the absence of Bgn and/or Dcn result in overall changes in tooth volume. 91 At least for Biglycan, and similar to the appendicular skeleton, these volumetric changes in the teeth are subjected to sexual dimorphism (different between males and females), which may explain the clinical observation of anomalous tooth morphology seen in certain X-linked diseases (since the Biglycan gene is only on the X chromosome). Biglycan is also known to play a role in the structures supporting the tooth—the periodontium, which comprises the gingiva, periodontal ligament (PDL), alveolar bone, and root cementum.17,122,123 The expression of Bgn in the PDL increases under physiologic load and tooth movement. 124 Examining Bgn/Fmod double-deficient mice revealed abnormal collagen fibrils in their PDL and altered remodeling of the alveolar bone. 17 During physiological root resorption of healthy human primary teeth, there is an upregulation in Biglycan expression in predentin and in pulp connective tissue, pointing to its involvement in regulating the resorption process. 117 Considering the prominent expression of Biglycan in many craniofacial tissues, it is not surprising that mutations in the human BGN gene cause varying degrees of craniofacial asymmetry and macrocephaly, as well as gingival hypertrophy. 125
The temporomandibular joint (TMJ) is a unique articulating joint in the craniofacial complex that is essential for proper mastication and speech. TMJ osteoarthritis is the most prevalent type of TMJ disorder. The loss of the cartilage and/or bone tissue results in permanent joint dysfunction and debilitating pain. Higher expression of BGN mRNA has been reported in samples from (human) patients with chronic TMJ diseases. 126 Both Bgn and Fmod are critical for the differentiation and function of TMJ chondrocytes by modulating TGF-β1 activity, 127 this is believed to be the etiology of the premature TMJ osteoarthritis that develops in Bgn/Fmod double deficient mice.
Human Genetics
The Biglycan gene is located on the X chromosome, and though it has neither a Y chromosome homolog nor an X inactivation escape mechanism, its expression level is different in patients with different numbers of sex chromosomes (e.g., Turner syndrome, 45, XO; Klinefelter syndrome, 47, XXY; Jacobs syndrome, 47, XYY, etc.). This and other evidence indicate “pseudoautosomal expression” of Biglycan may be attributed to a gene or genes that escape X inactivation and that regulate the transcriptional activity of Biglycan. 128
Recently, loss-of-function mutations in the human BGN gene were found to be responsible for Meester-Loeys syndrome (MRLS; OMIM: # 300989). 125 The identified mutations range from whole gene deletions to nonsense and splice site mutations, all resulting in loss of protein function. Fig. 3 depicts the locations and types of mutations found in the human BGN gene. Clinically, MRLS patients present with early-onset aortic aneurysm and dissection. The aortic phenotype in male BGN mutation carriers is typically severe and associated with high mortality rates, whereas female carriers range from unaffected to death due to aortic dissection. The males carrying the deletion also present with mild skeletal dysplasia, which is characterized by relatively short stature, phalangeal dysplasia, brachydactyly, hip dislocation, and dysplastic epiphyses of the long bones. There is considerable clinical overlap of MRLS syndrome with Marfan syndrome (OMIM: #154700) (e.g., arachnodactyly) and Loeys-Dietz syndrome (LDS, OMIM #609192) (e.g., hypertelorism, bifid uvula and cervical spine instability). Specific connective tissue findings such as pectus deformities, joint hypermobility/contractures, pes planus and striae, as well as malar hypoplasia, frontal bossing, proptosis/hypertelorism, down-slanting palpebral fissures, relative macrocephaly, and ventricular dilatation are also observed.
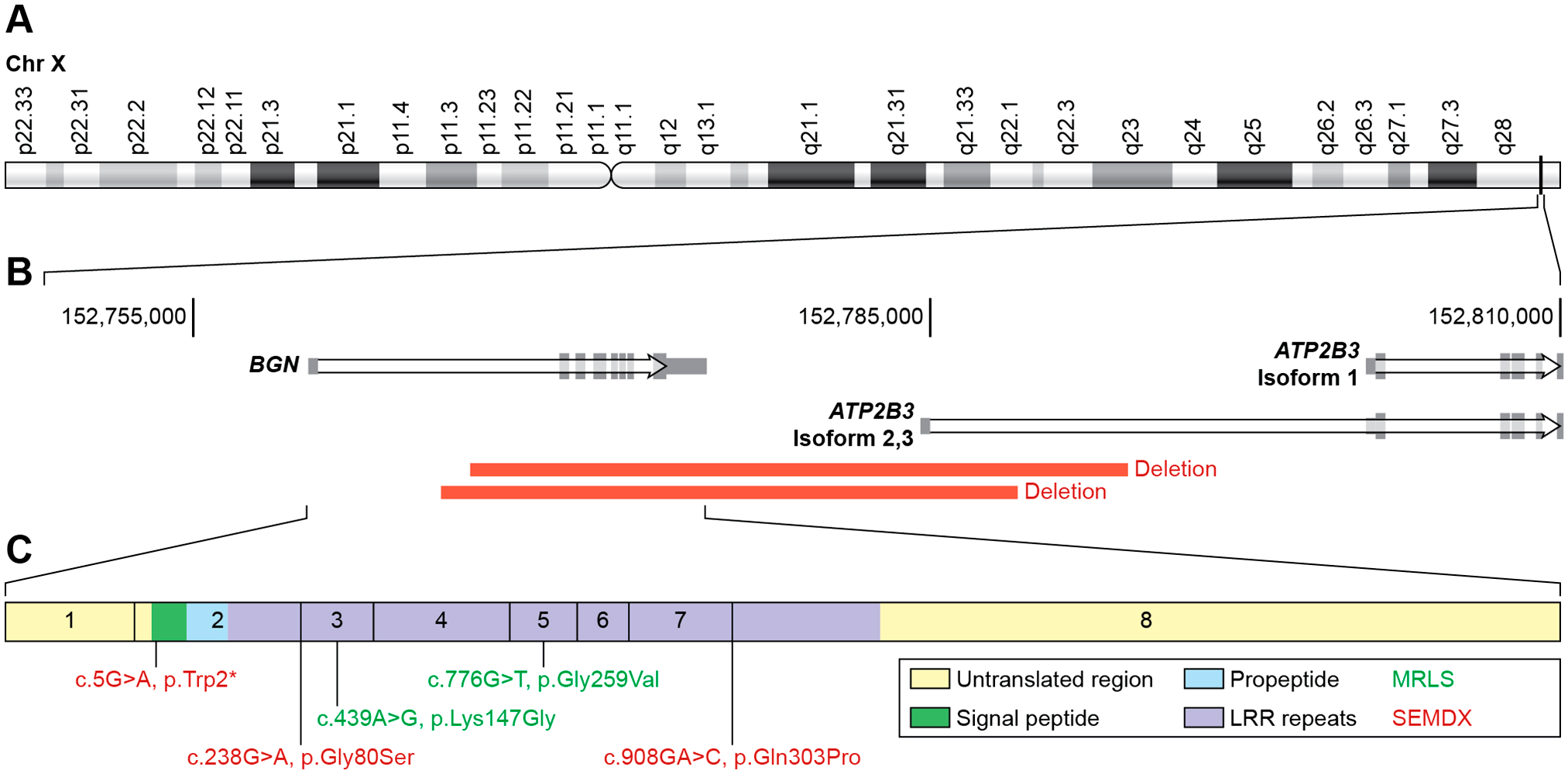
Published mutations in the BGN gene. (A) The location of Biglycan gene at the end of the long (q) arm of the human X chromosome. (B) Location of the current known deletions of the gene that cause Meester-Loeys syndrome. (C) High magnification of the exons of BGN illustrating the specific pathogenic variants found in human patients suffering from Meester-Loeys syndrome (in red) and X-linked spondyloepimetaphyseal dysplasia (in green). Abbreviations: BGN, Biglycan; LRR, leucine-rich repeat; MRLS, Meester-Loeys syndrome; SEMDX, spondyloepimetaphyseal dysplasia.
The molecular basis of MRLS was suggested to be increased TGF-β activity, as an increased number of pSMAD2 positive nuclei was observed in the aortic wall of patients. Importantly, increased TGF-β signaling has also been observed in aortic tissue of other thoracic aortic aneurysm and dissection syndromes, including Marfan and LDSs, and is considered the major contributor to most observed phenotypic features.125,129 The aortic phenotype is also observed in male mouse models of Bgn-deficiency, but only on a BALB/c background, which might be related to an inherent higher TGF-β sensitivity state. Structural abnormalities of the collagen fibrils were observed in these mice and could contribute to the weakness of the aortic wall.130,131
Interestingly, missense mutations that are not predicted to affect splicing events (p.Lys147Gly and p.Gly259Val) in the BGN gene, convey a different clinical presentation, namely spondyloepimetaphyseal dysplasia (SEMDX; OMIM #300106). The BGN-related SEMDX cases display epi- and metaphyseal changes in the long bones, leading to short stature, brachydactyly, bowing of the limbs, and waddling gate with lumbar lordosis. No cardiovascular anomalies were identified in these BGN mutation carriers.
It is unclear how BGN missense mutations lead to the development of SEMDX. Initially, the proposed disease mechanism was either impaired binding of TGF-β to the concave binding site of Biglycan or decreased Biglycan stability (leading to loss of function), but no functional evidence was provided for these hypotheses. 132 Many aspects of the pathogenetic mechanisms of Biglycan-related disease remain elusive and warrant further investigation.
Outlook
Despite its inert appearance, the skeleton is a very dynamic organ constantly undergoing remodeling. The multicellular unit—osteoblasts, osteocytes, and osteoclasts—regulates the matrix compartment, thereby maintaining optimal balance between the stiffness and flexibility of bones so that on the one hand they do not bend under changing (physiological) mechanical loading, but on the other hand remain flexible enough to dissipate the imparted energy to avoid microdamage or complete fracture. Moreover, being the body’s largest calcium-phosphate reservoir, the skeleton, through the remodeling process, is a key regulator of extracellular calcium and phosphate levels, governing bone-blood shifts of minerals.
PGs are major constituents of the ECM and in bone, SLRPs are the most abundant type of PGs. In addition to regulating collagen fibrillogenesis they also directly bind and interact with a large number of growth factors, morphogens, receptors, and other ECM structural and adhesive molecules, thus controlling the number and function of osteoblasts. Biglycan, a Type I SLRP, plays a crucial role in a multitude of physiological and pathological biological events such as cell attachment, proliferation and differentiation, morphogenesis, endochondral ossification, as well as tissue repair, inflammation, vascularization, bone remodeling, and fracture healing. While the expression of Biglycan in bone is well described, given the structural and functional significance of Biglycan, and SLRPs in general, in skeletal tissue architecture, development, and the effect on bone cells’ behavior, unraveling as much as possible of their biological role is central to our understanding of physiological and pathological processes. Though in recent years significant progress has been made in comprehending Biglycan’s function in the regulation of skeletal physiology, many unanswered questions remain including the following:
Does the expression of Biglycan in osteocytes influence mechanosensing in skeletal tissue?
Are there other, still undiscovered extracellular molecular partners that work in concert with Biglycan to control skeletal functions?
Does Biglycan’s pro-inflammatory role effect periosteal expansion and subsequent bone healing during fracture repair?
Do periosteal cells that make large amounts of Biglycan as the periosteal layer expands during the initial phases of fracture repair influence the healing process?
Why do loss-of-function mutations of BGN cause aortic aneurysms and dissections, while missense mutations in the same gene cause skeletal dysplasia?
As the world’s population steadily continues to age, osteoporosis, and osteoporotic fractures will constitute a growing public health burden. A better understanding of the cellular mechanisms of bone remodeling and the effects of different factors on these mechanisms, is likely to assist in identifying new targets for therapy that will assist in preventing and reversing bone fragility.
Footnotes
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
VK, RS, PJ, JANM, BL, and MFY all coordinated the writing, editing, and review. Each author has read and approved the manuscript prior to submission.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Work in the MFY’s laboratory is supported in part by the Intramural Research Program of the NIH, NIDCR, Molecular Biology of Bones and Teeth Section (Z01DE000379-35). Work in JM and BL’s laboratory is supported by Research Foundation—Flanders (FWO, 12X8520N). BL holds a consolidator grant from the European Research Council (Genomia—ERC-COG-2017-771945).