
Abstract
Airway epithelia play a crucial role in protecting the lung from the external environment. Ciliated airway epithelial cells contribute to mucociliary transport systems via ciliary beating and electrolyte transport mechanisms to defend against respiratory tract infection. Both of these activities are regulated by nitric oxide (NO)-dependent mechanisms. To better understand the role of the NO-cGMP signal transduction cascade in these responses, we investigated the localization of endothelial nitric oxide synthase (eNOS), soluble guanylyl cyclase (sGC), cGMP-dependent protein kinase (PKG) I-α, and PKG I-β in the tracheas and lungs of normal rats by immunohistochemistry. Mouse anti-eNOS, rabbit anti-sGC, PKG I-α, and PKG I-β antibodies were used. Strong immunostaining for eNOS was detected in ciliated tracheal, bronchial, and bronchiolar epithelia, in Clara cells, and in Type II alveolar cells. The pattern of sGC and PKG I-β immunostaining showed striking parallels with that of eNOS staining. No staining was detectable in ciliated epithelium with the anti-PKG I-α antibody. Taken together, these observations suggest that PKG I-β might transduce NO-sGC signaling into biological responses in ciliated respiratory epithelia.
Keywords
NITROUS OXIDE (NO) a physiologically important activator of sGC, plays an important regulatory role in airway function and is implicated in pulmonary physiology. There is evidence that ciliary beating and electrolyte transport are regulated by NO-dependent mechanisms (Jain et al. 1993,1995; Takemura et al. 1995) and that eNOS is found in ciliated epithelia of rat lung and is specifically localized to the basal body of the microtubule of the cilia (Xue et al. 1996). Furthermore, low levels of nasal NO correlate with impaired mucociliary function in the upper airway (Lindberg et al. 1997), and NOS inhibitors decrease ciliary beating frequency (CBF) after prestimulation with isoproterenol, bradykinin, or substance P (Jain et al. 1993), indicating a novel NO-dependent mechanism that upregulates ciliary motility in response to stimulation. The role of cGMP as a mediator of NO signaling has recently focused attention on another area of investigation, the role of PKG and other potential cGMP receptor proteins in cell function. Surprisingly little is known about the mechanisms by which cGMP mediates the actions of NO. Both kinase-dependent and -independent actions of cGMP have been reported. The localization of PKG isoforms has been reported in kidney and other tissues (Gambaryan et al. 1996; Pryzwansky et al. 1995), but little is known of the localization of PKG in ciliated epithelium. Elucidating the presence and localization of PKG in ciliated epithelium would provide important information for understanding the signal transduction pathway for NO regulation. To better appreciate the “downstream” effects of NO cGMP signaling mechanisms in the lower airway and ciliated epithelium, we examined and localized eNOS, sGC, PKG I-α, and PKG I-β protein expression in rat trachea and lung tissues.
Materials and Methods
Tissue Preparations
All rats were obtained from Hilltop Laboratory Animals (Scottdale, PA) and were treated in accordance with APS/NIH guidelines. Six normal male Sprague-Dawley rats were anesthetized with isoflurane. Their lungs and tracheas were rapidly removed and cut as cross-sections approximately 2 mm thick. The specimens were then immersed in fixative solution containing 4% paraformaldehyde in PBS (0.1 M, pH 7.4). After 3 hr of fixation, the specimens were dehydrated in graded ethanol for paraffin embedding. Sections of 5 μm were cut (at least six slides for each lung) and mounted on Superfrost/Plus (Fisher; Pittsburgh, PA) slides for immunostaining.
Western Blot Analysis
To examine the eNOS antibody specificity, Western blot analysis was performed. For the preparation of crude extracts, rat lung, brain, spinal cord tissues, and rat lung tissues induced with LPS (10 mg/kg body weight IP for 6 hr) were homogenized in ice-cold 50 mM Tris-HCl, pH 7.4, containing 0.1 mM EDTA, 0.1 mM EGTA, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), 2 mM leupeptin, 1 mM pepstatin, and 0.1% 2-mercaptoethanol. The homogenate was centrifuged at 15,000 X g for 30 min at 4C and the pellet was discarded. The protein of the supernatant was measured using a Bio-Rad kit according to the manufacturer's instructions (Bio-Rad; Hercules, CA). The samples of lung, brain, and spinal cord, were loaded (100 x μg each) and separated on a 7.5% SDS-PAGE, followed by blotting of the proteins to nitrocellulose membrane (Bio-Rad). The blot was blocked with a buffer consisting of 50 mM Tris-HCl, pH 7.4, 0.15 M NaCl, 2% nonfat milk, 2% bovine serum albumin, and 0.1% Tween-20, for 1 hr at room temperature (RT). The blot was then incubated with monoclonal mouse anti-eNOS antiserum (1:500 dilution; Transduction, Lexington, KY) for 1 hr at RT, followed by a secondary goat anti-mouse IgG conjugated with horseradish peroxidase (HRP) (Bio-Rad), and detected with ECL blotting detection reagents (Amersham; Poole, UK). After detection, the film was quantitated with a densitometer and Imagequant software (Molecular Dynamics; Sunnyvale, CA).
Antibodies for Immunohistochemistry
A mouse monoclonal antibody against eNOS was obtained from Transduction (1:500 dilution). Rabbit polyclonal antibodies against sGC were produced using synthetic peptide from the α1- and β1-subunits of sGC (Nakane et al. 1990) (1:500 dilution; Cayman Chemical, Ann Arbor, MI). PKG I-α was made from a 15-residue synthetic peptide based on the human PKG I-α (residues 657-671) and PKG I-β from a 14-residue synthetic peptide based on the human PKG I-β (Sandberg et al. 1989) (residues 4-17; 1:500 dilution for both PKG I-α and PKG I-β; StressGen, Victoria, BC, Canada). The antibodies were used for detection of eNOS, sGC, PKG I-α, and PKG I-β, respectively.
Immunohistochemistry
After deparaffinizing, the slides were treated with 3% H2O2 in PBS for 20 min to quench endogenous peroxidase activity. After blocking of nonspecific sites with 10% goat serum in PBS for 60 min, tissue sections were washed (three times for 5 min in PBS) and incubated with specific antibody at 4C overnight. After washing off unbound primary antibodies with PBS, the sections were incubated with biotinylated anti-mouse (eNOS) or anti-rabbit (sGC, PKG I-α, and PKG I-β) serum (1:200 dilution) for 1 hr. Specific binding was detected using an avidin-biotin-HRP complex (1:100 dilution for 1 hr; Vector, Burlingame, CA) and a substrate solution of H2O2 and diaminobenzidine (DAB) kit (Vector) according to the manufacturer's instructions. Then the slides were counterstained with hematoxylin, dehydrated through graded alcohol and xylenes, mounted on coverslips, and examined under an Olympus VANOX AHBS3 microscope. Negative controls were carried out with normal rabbit serum and mouse IgG according to the primary antibodies used for sGC, PKG I-α, PKG I-β, and eNOS staining.
Results
Immunospecificity of eNOS Antibodies
In Western blot (Figure 1A), eNOS antibody was shown to react only with eNOS protein at 135-kD mass in rat lung (Lane 1 in Figure 1A), spinal cord (Lane 3 in Figure 1A), and LPS-induced lung (Lane 4 in Figure 1A), but not in the rat brain (Lane 2 in Figure 1A). The antibody did not crossreact with bNOS and iNOS proteins because there were no proteins at 155-kD mass and 130-kD mass (the kD mass of bNOS and iNOS). The result from Western blotting indicated that the eNOS antibody was specific for the eNOS protein and did not detect other NOS isoforms in rat tissues.
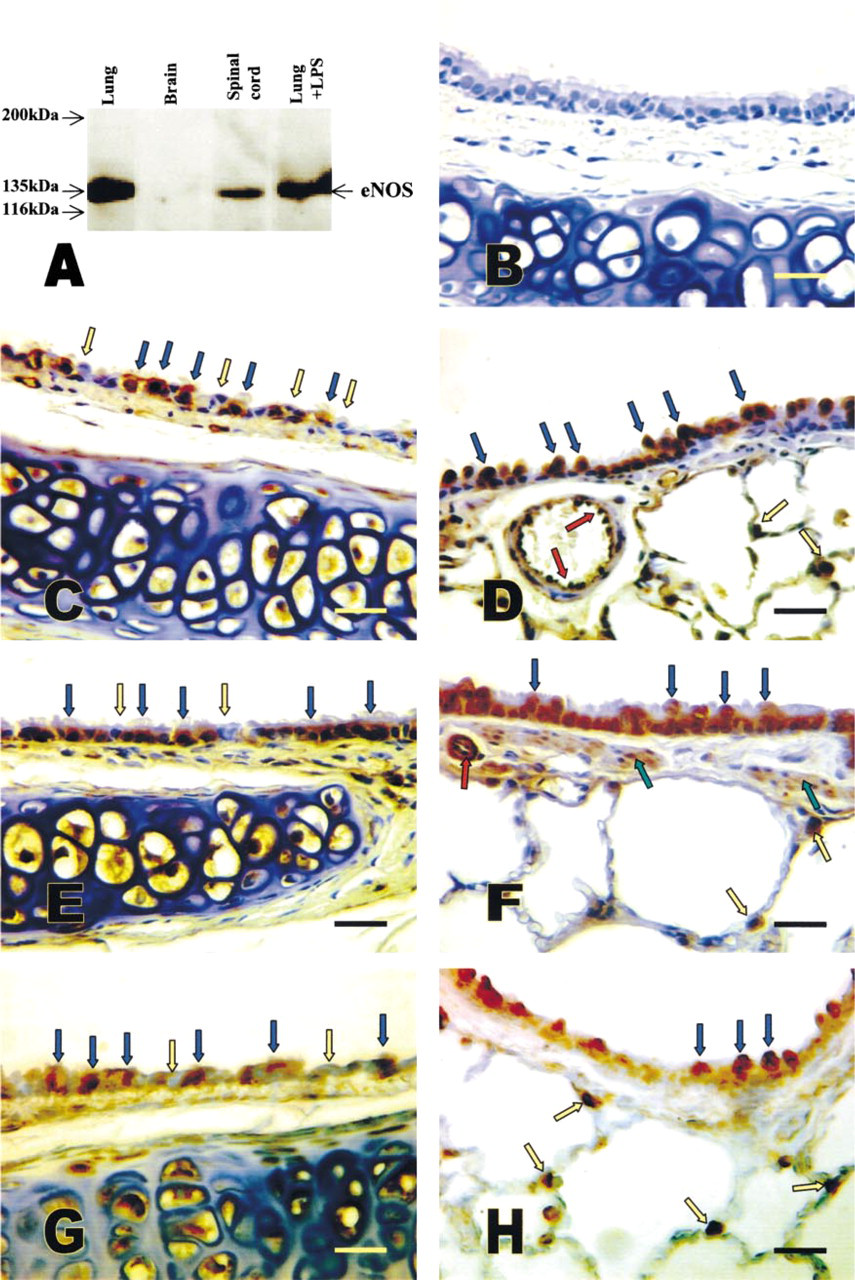
Western blot of eNOS from different organs and immunohistochemical localization of eNOS, sGC, and PKG I-β in the airways. (A) Western blot showing the specific immunoreactivity of eNOS antibody at 135-kD mass in rat lung (Lane 1), spinal cord (Lane 3), and LPS-induced lung (Lane 4) but not in the brain (Lane 2). (B) Negative control section immunostained with mouse IgG. eNOS (C,D) immunostaining was detected in ciliated epithelium in trachea (blue arrows in C), Clara cells (blue arrows in D), on endothelial cell of a bronchial artery (red arrows in D), and Type II alveolar cells (yellow arrows in D). A similar sGC (E,F) and PKG I-β (G,H) staining pattern was also shown in the ciliated epithelium (blue arrows in E and G), Clara cells (blue arrows in F and H), and alveolar Type II cells (yellow arrows in F and H). Bars = 25 μm.
Immunoreactivity of eNOS, sGC, PKG I-α, and PKG I-β in Lung and Trachea
To determine the distribution of eNOS, sGC, PKG I-α, and PKG I-β in lung and trachea, we performed immunohistochemical analysis in sections. With eNOS staining of tracheal and bronchial tissues, we detected a strong and specific signal in ciliated epithelium (Figure 1C, blue arrows; trachea), with prominent staining of eNOS in Clara cells of the bronchioles (Figure 1D, blue arrows) and in Type II cells of alveoli (Figure 1D, yellow arrows). Nonciliated epithelia in trachea and bronchus (Figure 1C, yellow arrows) were not stained with eNOS. In conduit vessels, the endothelial cells were strongly positive for eNOS staining (red arrows in Figure 1D). The pattern of sGC (Figures 1E and 1F) and PKG I-β (Figures 1G and 1H) immunostaining showed striking parallels with that of eNOS immunostaining in ciliated epithelia in trachea and bronchus (Figures 1E and 1G, blue arrows; bronchi), in the Clara cells of the bronchioles (Figures 1F and 1H, blue arrows), and in Type II cells of alveoli (Figures 1F and 1H, yellow arrows). Moreover, PKG I-β was localized in the smooth muscle of larger vessels and sGC was localized in smooth muscle of both large and small vessels (red arrows in Figure 1F) as well as airway smooth muscle (green arrows in Figure 1F). Nonciliated epithelia in trachea and bronchus were stained neither with sGC (Figure 1E, yellow arrows) nor with PKG I-β (Figure 1G, yellow arrows) antibody. Negative controls showed no background staining (Figure 1B, for eNOS control). In contrast to PKG I-β, staining with the anti-PKG I-α antibody revealed a strong signal in the smooth muscle of airway and resistance vessels. No staining was detectable in ciliated epithelium with anti-PKG I-β antibody (Figure 2). All the immunohistochemical staining for each antibody was performed at least three times in consecutive sections from the six animals. The staining pattern and intensity for each antibody showed very little variation among the animals and in different sections. To summarize, immunostaining for eNOS, sGC, and PKG I-β was positive in all regions of the respiratory tract of trachea and lung, whereas PKG 1-α immunostaining was negative throughout the epithelium of the airways.
Discussion
NO, derived from L-arginine by NOS, is involved in regulation of several important physiological functions. Previous studies have demonstrated that eNOS is localized in ciliated epithelium of rat lung (Xue et al. 1996). Direct evidence for the presence and distribution of the downstream components of the NO signaling pathway, sGC, and PKG in ciliated epithelia of airway have not been described completely. To address this issue, we sought to identify the anatomic and cellular sites of the NO-sGC signal pathway in lung and trachea. The findings indicate that many specific sites of eNOS, sGC, PKG I-α, and PKG I-β immunostaining exist in trachea and lung. Specifically, sGC and PKG I-β accompanied eNOS immunostaining in all ciliated epithelia, consistent with a role of the NO-cGMP pathway in regulating ciliary beat frequency. A previous study reported by our laboratory (Rengasamy et al. 1994) demonstrated that sGC is present only in the smooth muscle cells of the airways and is not found in the epithelial cells of the airways. This discrepancy may result from the antibodies used in these two studies. In the previous study, a monoclonal anti-sGC 72-kDa β1-subunit antibody was used (Brandwein et al. 1981). In the present study, a polyclonal rabbit anti-sGC antiserum was applied. This antiserum can react with both α1- and β1-subunits of sGC. The polyclonal anti-sGC antiserum may be more sensitive than the monoclonal antibody and may react with the sGC α1-subunit present in the airway epithelial cells.
In vitro studies have shown that several mediators or transmitters such as isoproterenol, substance P, bradykinin, TNF-α, and IL-1β employ an NO-dependent mechanism to upregulate ciliary motility (Jain et al. 1993,1995). NG-Monomethyl-L-arginine (L-NMMA), a NOS inhibitor, prevented CBF stimulation effects of these ciliary stimulators in bovine airway ciliated epithelium. This reduction of CBF induced by NOS inhibition was completely reversed by L-arginine or sodium nitroprusside (SNP) but not by D-arginine. These studies demonstrated a novel NO-dependent mechanism that upregulates ciliary motility in response to stimulation. Our findings provide direct evidence supporting the view that NO is an important regulator of ciliary function in the airway.
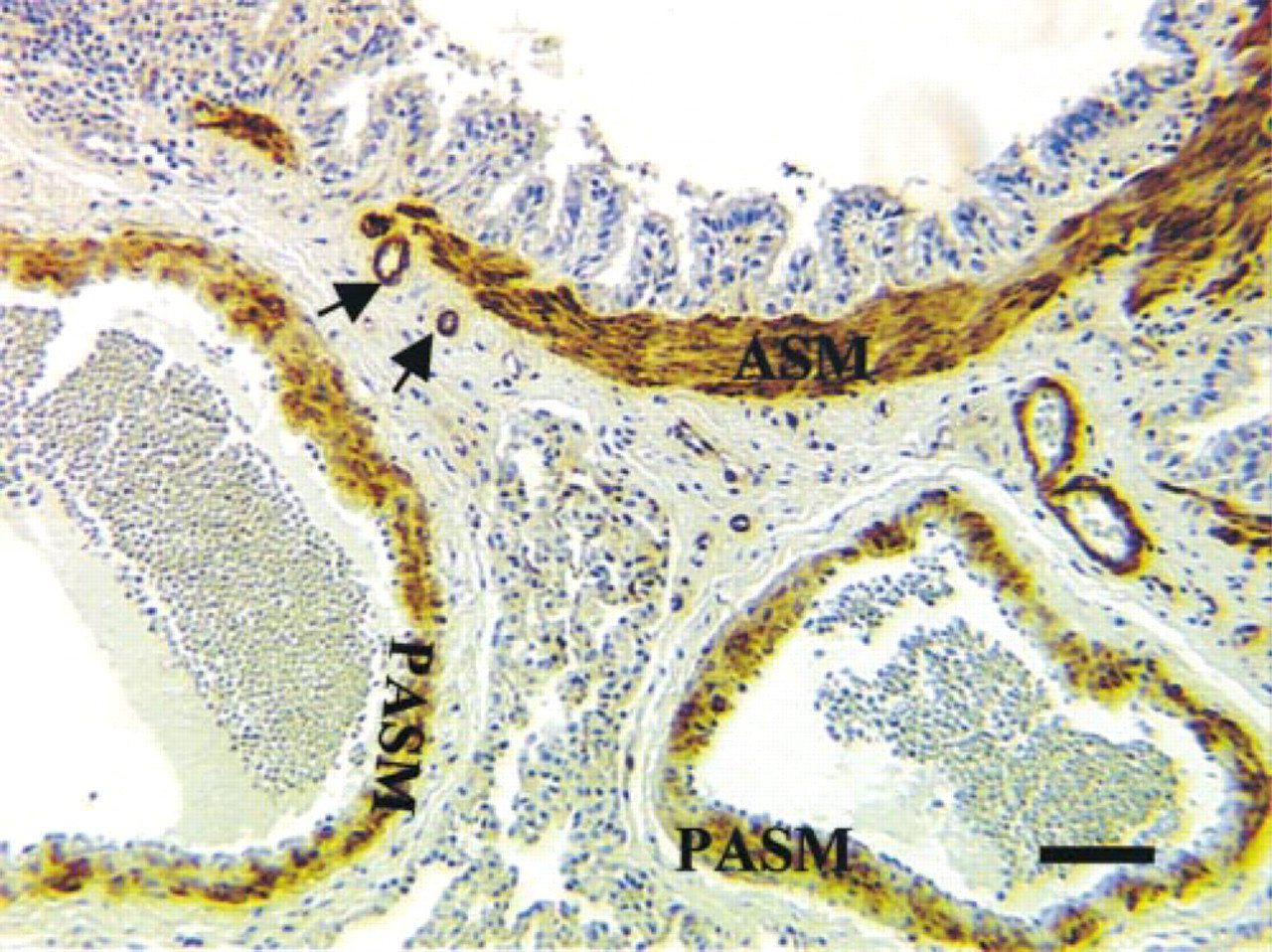
Immunostaining of PKG I-α in rat lung. Intense immunopositivity of PKG I-α is present in the airway smooth muscle and smooth muscle of bronchial artery (arrows). The pulmonary artery smooth muscle showed relatively weaker staining for PKG I-α compared to airway and bronchial artery smooth muscle. Bar = 50 μm.
If cGMP acts as a second messenger in this tissue, it is likely to require the presence of a PKG together with the presence of appropriate enzymes to degrade the signal. In this context, Yang et al. made an important in vitro observation about the effect of methacholine on the CBF of human adenoid explants (Yang et al. 1997). They demonstrated the roles of endogenous prostaglandins in methacholine-induced ciliostimulation by treating specimens with a cyclo-oxygenase inhibitor (diclofenac sodium). Diclofenac significantly inhibited the ciliostimulatory effects of methacholine. NG-nitro-L-arginine-methyl ester (L-NAME), an NOS inhibitor, inhibited the effects of methacholine in L-arginine-free minimal essential medium, and this inhibition was reversed by L-arginine. Furthermore, they found that a cGMP inhibitor, KT-5823, significantly inhibited the effects of methacholine. Their investigation suggests that cilia stimulation by methacholine in human upper airway mucosa involves both prostaglandin and NO second messengers as well as activation of PKG. In this report we demonstrate for the first time that the PKG I-β isoform is present in rat ciliated airway epithelium. PKG I-β was localized to the same sites as eNOS and sGC in epithelium of lung, suggesting that PKG I-β might transduce NO-cGMP signaling into biological responses in pulmonary epithelium.
It is widely assumed that NO diffuses passively from the cytosol of its cell of origin into distant target cells to elicit a response (Gaston et al. 1994). However, others have shown that NO is concentrated on the surface of porcine endothelial cells (Malinski and Taha 1992) and that NO does not pass as freely into pulmonary arterial media from the adventitial side as from the intimal side (Steinhorn et al. 1994), suggesting that NO from vascular tissue is not readily diffusible into the airway. Helium embolization of the rabbit pulmonary artery does not affect exhaled NO concentrations (Gustafsson et al. 1991) and alveolar gas NO is significantly less than in upper airways and bronchioles (Dweik et al. 1998), suggesting an NO source within the airway independent of the vasculature. NO in the airway may originate from cells in the airway wall, including epithelial cells, mast cells, or neurons. Our findings confirm that ciliated epithelia in airway are a likely source of exhaled NO.
The physiological actions of NO-cGMP in vasculature have been very well characterized (Murad 1994) but the functions of the NO-cGMP and PKG I-β in respiratory epithelium have not yet been elucidated. Their role in modulation of cilia motility is still unclear. Recent studies demonstrated that the NO-cGMP signal transduction pathway may stimulate CBF by influencing the intracellular calcium concentration (Uzlaner and Priel 1999). However, the exact mechanism of how NO-cGMP and PKG I regulate CBF still needs to be clarified. Biological motion dependent on ATP hydrolysis is often generated by two distinct groups of proteins: actin and its ATPase myosin, and tubulin and its ATPase dynein (Warner and Mitchell 1980). Just as myosin provides the motive force in the smooth muscle, dynein provides the motive force in the cilia. The motion of cilia is achieved by coupling the hydrolysis of ATP by dynein to micro-tubule sliding (Walczak and Nelson 1994). It is well established that cGMP activates PKG and alters phosphorylation of many endogenous proteins, including an unidentified protein that might lead to the dephosphorylation of the myosin light chain and relaxation (Murad 1994). In cilia, both cAMP and cGMP increase dynein-like ATPase activity (Walczak and Nelson 1994). Satir and Matsuoka (1989) found that dynein arms of half of the microtubule doublets in metazoan cilia were active during the effective stroke and that the activity of these arms was diminished during the recovery stroke, when the activity of the other half of the dynein arms was turned on. Therefore, it appears that the enhancement of CBF by cAMP or cGMP in the cilia may be due to an increase in the rate of relaxation. A recent study reported by Wyatt et al. (1998) demonstrated that both cAMP-dependent protein kinase (PKA) and PKG are involved in the regulation of CBF in bovine bronchial epithelial cells (BBECs). cAMP levels and PKA activity are increased in BBECs stimulated with 0.01 mM isoproterenol, with a corresponding increase in CBF. Cyclic GMP levels and PKG activity are increased in BBECs stimulated with 0.1-10 μm sodium nitroprusside, with a corresponding increase in CBF. Direct protein kinase-activating analogues of cAMP and cGMP also activated their specific kinase and stimulated CBF. Preincubation of BBECs with inhibitors of PKA or PKG results in the inhibition of specific kinase activity as well as in the inhibition of CBF. This study support our findings, which suggest that NO-cGMP-PKG regulates airway epithelial CBF.
A special type of human affliction called Kartagener's syndrome has been shown to result from absence of dynein arms in the cilia (Palmblad et al. 1984) and to correlate with the absence of NO in nasal air (Lundberg et al. 1994). Therefore, this syndrome may have a multifactorial pathophysiology. It is interesting to speculate that there may be an interaction between NO and dynein. Dynein has been shown to be homologous with PIN, an endogenous associated protein inhibitor of neuronal nitric oxide synthase (Jaffrey and Snyder 1996).
In summary, the finding of strong immunostaining for eNOS, sGC, and PKG I-β in airway ciliated epithelia suggests an important role for NO-cGMP in the physiology of the airway and that PKG I-β is available to transduce NO-sGC signaling into biological responses in ciliated airway epithelium. In contrast, PKG I-α was localized primarily in smooth muscle. Therefore, we speculate that each isoform of PKG I may be closely linked with its own unique function in the lung.
Footnotes
Acknowledgements
Supported by NIH grants R01 HL 39706 and R01-GM 49111.