
Abstract
A radioactive and biotin-labeled analogue of GM1 (biotin-GM1) was synthesized which enabled us to analyze its intracellular distribution in the compartments of the endocytic route by electron microscopic immunocytochemistry using thin sections of human skin fibroblasts labeled with gold-conjugated antibiotin antibodies. Metabolic studies with the biotin-GM1 showed its partial degradation to the corresponding GM2 and GM3 derivatives. Further degradation was inhibited by the biotin residue. The distribution of biotin-GM1 after uptake by cells was studied by postembedding labeling techniques. On the plasma membrane the biotin-GM1 was detectable in the form of patches (0.1 μm in diameter), in caveola-like structures and, to a much lesser extent, in coated pits or vesicles. During endocytic uptake, the biotin-GM1 became detectable in organelles identified as late endosomes and lysosomes. The intracellular distribution of the biotin-GM1 was compared to the localization of the EGF receptor in EGF-stimulated fibroblasts. Both the biotin-GM1 and the EGF receptor were transported to intraendosomal and intralysosomal membranes, indicating that both membrane constituents follow the same pathway of endocytosis. Our observations show that biotin-GM1 can be successfully incorporated into the plasma membrane and be used as a tool for morphological detection of its pathway to lysosomes. (J Histochem Cytochem 47:1005–1014, 1999)
G
Although biosynthesis and degradation of gangliosides are biochemically well characterized (Schwarzmann and Sandhoff 1990; van Echten and Sandhoff 1993), much less is known about the intracellular transport of gangliosides in living cells. Morphological studies on the intracellular trafficking of membrane lipids often depended on fluorescent lipid analogues (van Meer 1989; Pagano 1990; Pütz and Schwarzmann 1995; Sofer et al. 1996) and were restricted to the limited resolution of light microscopy. Electron microscopic studies employed cholera toxin as a probe for the localization of GM1 (Hansson et al. 1977; Parton 1994) or antibodies against Forssman glycolipid (van Genderen et al. 1991). These studies offer important insights into the localization of endogenous glycosphingolipids. For transport studies, however, labeled lipid derivatives are required that can be distinguished from the endogenous pool of lipids. The major advantage of a tagged exogenous ganglioside is the specificity of the antibody recognition of the tag. This gives unambiguous results in contrast to the use of anti-ganglioside antibodies (Schwarz and Futerman 1997).
Because it is possible to insert exogenous gangliosides into the plasma membrane of cultured cells (Callies et al. 1977; Radsak et al. 1982; Schwarzmann et al. 1983, 1987; reviewed by Saqr et al. 1991), ganglioside analogues could be used to visualize endocytic membrane flow. For this purpose we synthesized a biotin-tagged GM1 derivative (biotin-GM1; Albrecht et al. 1997) to study its intracellular transport along the endocytic pathway by electron microscopy using gold-conjugated anti-biotin antibodies. Another important prerequisite for visualization is the resistance of the biotin label to degradation by cells. This was confirmed before using a [14C]-labeled biotin-GM1 for in vitro degradation and metabolic studies. The biotin-GM1 was shown to be resistant to the action of sialidase but was characteristically degraded to the corresponding GM2 and GM3 derivatives. Further degradation is inhibited by the biotin residue (Albrecht et al. 1997). This makes the biotin-GM1 a valuable tool for studies of the endocytic membrane flow because the metabolic products show that biotin-GM1 reached degradative compartments. Even after partial degradation the molecule remains a biotin-labeled ganglioside incapable of spontaneous transbilayer movement (flip-flop) and therefore restricted to vesicular transport. The major technical challenge of this study was the preservation of the ultrastructure while simultaneously localizing biotin-GM1.
Immunocytochemistry of proteins is a well-established and extensively studied field (Hayat 1989; Griffiths 1993), whereas very few studies exist on lipid immunocytochemistry on the EM level (Schwarzmann et al. 1987; van Genderen et al. 1991; Parton 1994; Kobayashi et al. 1998). One reason may be the lack of well-characterized fixatives for lipids, in contrast to a broad variety of fixatives for proteins. For determination of the subcellular distribution of biotin-GM1, we used two different approaches to avoid the problem of lipid redistribution: plastic embedding in LR Gold of OsO4-postfixed samples after a progressive lowering of temperature (PLT) protocol and a modified cryosectioning protocol for improved structural preservation (Liou et al. 1996).
In this study we examined the intracellular distribution of biotin-GM1 that participated for many hours in the endocytic membrane flow. Because the exogenous gangliosides and their derivatives, such as bi-otin-GM1, become components of the plasma membrane, they can be used as membrane tracers for electron microscopy. Moreover, the technique developed in the course of this study allows direct visualization at high spatial resolution of the distribution of individual biotin-labeled GM1 molecules.
We were able to show the incorporation of biotin-GM1 into the plasma membrane, where it became detectable mainly in circumscribed patches and in caveola-like structures. Because late endosomes and lysosomes contain internal membrane structures in the form of vesicles or lipid lamellae (Hopkins et al. 1990; van Deurs et al. 1995; Mellman 1996) it was of particular interest to determine whether the biotin-GM1 would be sorted to these internal membranes in the course of endocytosis, the likely site of glycosphin-golipid catabolism (Fürst and Sandhoff 1992). The results show that, on endocytosis, biotin-GM1 was detectable over intraendosomal and intralysosomal membranes where it met with peptide markers of endocytosis, e.g., the EGF-EGF receptor complex.
Materials and Methods
All mixtures of solvents are given in proportions of volumes, and this is therefore not further indicated.
Cell Culture
Monolayer cultures of human skin fibroblasts obtained from biopsies of a male infant were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, Eggenstein, Germany) supplemented with 10% (v/v) fetal calf serum (FCS; Cytogen, Berlin, Germany). Cells were grown at 37C in a water-saturated atmosphere of 5% CO2 in air.
Antibodies
The following mouse monoclonal antibodies (MAbs) were used in this study: H4A3, which reacts with LAMP-1, and H5C6, which reacts with LIMP (both from Developmental Studies Hybridoma Bank; Baltimore, MD); and EGF-R1, which reacts with an extracellular domain of the EGF receptor (Waterfield et al. 1982; obtained from Biotrend, Köln, Germany). The following polyclonal antibodies were used: anti-biotin antibodies, developed in goat, conjugated to ultra-small gold particles (GP-US) or 10-nm gold particles (Aurion; Wageningen, The Netherlands); anti-MPR antibodies, developed in rabbit (provided by Dr. Bernd Hoflack; Institut Pasteur, Lille, France) and anti-caveolin-1 antibodies, developed in rabbit (Santa Cruz Biotechnology; Heidelberg, Germany) were also used. Gold-conjugated (6 nm) goat anti-mouse and goat anti-rabbit antibodies were obtained from Aurion. For immunolabeling, antibodies were diluted in 0.2 M HEPES, pH 7.2, containing 1% BSA and 0.2% CWFS-gelatin.
Analysis of Biotin-GM1 Uptake and Metabolism in Cultured Fibroblasts
Incubation media containing biotin-GM1 were prepared as described previously (Albrecht et al. 1997). Fibroblasts grown to confluency in 25-cm2 tissue culture flasks were washed in DME and incubated with 10 μM of biotin-GM1 in DME containing 0.3% FCS for 72 hr and 120 hr at 37C. The presence of 0.3% FCS in the biotin-GM1-containing culture media was chosen as a compromise between cell viability and inhibition of ganglioside uptake by cells because of ganglioside adsorption to serum proteins (Sonderfeld et al. 1985). The incubation medium was removed, cells were thoroughly washed with PBS, and then were harvested with a solution of trypsin/EDTA in PBS (1% trypsin, 0.002% EDTA) (Radsak et al. 1982). The trypsinized cells were collected by centrifugation. After washing with cold PBS, the resulting cell pellet was resuspended in 800 μl water and an aliquot of 5 μl was analyzed for protein as described by Bradford (1976), using BSA as standard. For lipid extraction, the remainder of the cell suspension was treated with chloroform/methanol (1:2) for 48 hr at 37C. After evaporation in a stream of argon and desalting with LiChroprep RP18 (Merck; Darmstadt, Germany), lipid extracts were dissolved in 1 ml chloroform/methanol (1:1) and an aliquot of 10 μl was taken to determine radioactivity by a liquid scintillation counter Tri-Carb 1900 (Canberra Packard; Frankfurt, Germany), using Ultima Gold (Canberra Packard) as scintillation cocktail. The lipids were separated by TLC (chloroform/methanol/15 mM calcium chloride, 60:35:8) and the radioactive bands visualized on X-ray-sensitive film.
PLT Embedding in LR Gold and Immunolabeling
Human skin fibroblasts grown in 25-cm2 flasks were incubated with biotin-GM1 as described above for 72 hr at 37C. In controls, the biotin-GM1 was omitted. Cells were harvested by treatment with a solution of proteinase K (Merck) 0.05 mg/ml in PBS for 3 min on ice, pelleted, fixed with 4% formaldehyde in 0.2 M HEPES, pH 7.4, and postfixed with 1% OsO4 for 10 min at 4C. In control samples, OsO4 fixation was omitted. Pellets were embedded in LR Gold (Poly-sciences; Warrington, PA) after a progressive lowering of temperature (PLT) embedding protocol: dehydration in ethanol 50% for 45 min at 0C, 70% for 60 min at −20C, 90% for 60 min at −20C; infiltration in LR Gold/ethanol 1:1 for 30 min at −20C, 7:3 for 60 min at −20C, pure LR Gold for 60 min at −20C, LR Gold containing 0.5% benzil at −20C for 60 min, overnight, and for 1 hr. Polymerization was achieved with UV light for 48 hr at −20C and for 48 hr at room temperature. Immunolabeling of sections was performed with goat anti-biotin antibodies conjugated to 10-nm gold particles (1:100), co-incubated with mouse anti-LAMP-1 MAb (H4A3, 1:100), followed by 6-nm goat anti-mouse-gold (1:50), or co-incubated with rabbit anti-MPR antibodies (1:120), followed by 6-nm goat anti-rabbit-gold (1:50). Controls included single labeling for each antibody and deletion of the primary antibody. Sections were postfixed with 2% glutaraldehyde in 0.2 M HEPES, stained with 2% uranyl acetate, and viewed at 80 kV with a Philips CM 120 electron microscope (Philips; Eindhoven, The Netherlands).
Cryoultramicrotomy and Immunolabeling
Human skin fibroblasts grown in 25-cm2 flasks were incubated with biotin-GM1 as described above for 72 hr at 37C. For controls, biotin-GM1 was omitted. Cells were harvested with proteinase K as described above, pelleted, and fixed with 4% formaldehyde and 0.1% glutaraldehyde in 0.2 M HEPES, pH 7.2. Pellets were infused with 50% polyvinylpyrrolidone (PVP-10,000; Sigma, Deisenhofen, Germany) and 1.15 M sucrose in 0.1 M HEPES (modified according to Tokuyasu 1989) at 4C overnight on a rotator, placed on specimen holders, and frozen in liquid nitrogen. Cryosections were collected in a 1:1 mixture of 2% methylcellulose (Sigma; 2% = 25 cps, 25C) and 2.3 M sucrose according to Liou et al. (1996). Thawed sections were washed with distilled water before immunolabeling. Sections were immunolabeled with goat anti-biotin antibodies coupled to 10-nm gold particles (1:100, 1 hr). Double labeling experiments were performed by co-incubation with anti-biotin antibodies, conjugated to 10-nm gold particles, and mouse anti-LIMP MAbs (1:40, 1 hr) or rabbit anti-caveolin-1 antibodies (1:100, 1 hr), followed by goat anti-mouse conjugated to 6-nm gold particles (1:50, 1 hr) or by goat anti-rabbit conjugated to 6-nm gold particles (1:50, 1 hr). Controls included single labeling for each antibody and deletion of the first antibody. After postfixation with 2% glutaraldehyde in 0.2 M HEPES, pH 7.2, sections were embedded in 1.8% methylcellulose containing 0.4% uranyl acetate according to Griffiths et al. (1984) and were viewed at 80 kV with a Philips CM 120 electron microscope.
Localization of the Biotin-labeled GM1 Analogue in Comparison to the EGF Receptor in EGF-stimulated Fibroblasts
Fibroblasts were incubated for 72 hr at 37C with biotin-labeled GM1 analogues at a concentration of 10 μM in DME containing 0.3% FCS. Cells were washed thoroughly and incubated with 100 nM EGF (Sigma) for 1 hr at 4C according to Beguinot et al. (1984). After the cold medium was replaced by warm DMEM containing 100 nM EGF and 10% FCS, cells were further incubated for 45 min at 37C and then harvested with proteinase K. After the cells had been pelleted, they were fixed with 4% formaldehyde and 0.1% glutaraldehyde in 0.2 M HEPES and processed for LR Gold embedding or cryosections. In further experiments, cells were incubated for 120 min at 37C with 100 nM EGF before processing as above to allow EGF internalization and downregulation of EGF receptors. These experiments served as controls. Sections were labeled as described by co-incubation with goat anti-biotin antibodies coupled to 10-nm gold particles and mouse MAb EGF-R1 (1:5, 1 hr), followed by incubation with goat anti-mouse antibodies coupled to 6-nm gold particles (1:50, 1 hr). Controls included single labeling and deletion of the primary antibody.
Results
Uptake and Metabolism of Biotin-GM1 by Human Fibroblasts
For biochemical studies, fibroblasts were incubated with 10 μM biotin-GM1 (Figure 1) in DME with 0.3% FCS for 72 hr and 120 hr at 37C. The same concentration of biotin-GM1 was used for morphological studies. After careful washes, the cells were harvested as described in Materials and Methods and the lipids were extracted from the cell pellets. The amount of cell-associated biotin-GM1 was 4.4 ± 0.5 nmol/mg protein at both time points, indicating saturation of ganglioside uptake. For analysis of metabolic products of biotin-GM1, the extracted lipids were separated by TLC and radioactive lipids were visualized by exposure to X-ray-sensitive film. As shown in Figure 2, biotin-GM1 has been degraded to the corresponding GM2 and GM3 derivatives. Their structures have been confirmed previously by digestion of biotin-GM1 with the appropriate glycohydrolases (Albrecht et al. 1997). Below the biotin-GM1 and biotin-GM2 bands, three weak radioactive bands appeared (Figure 2, × 1, × 2, × 3) representing the sulfoxides of the respective biotin gangliosides. These results agree well with the previous study carried out with lower concentrations of biotin-GM1 (Albrecht et al. 1997). Under the conditions employed, biotin-GM1 was degraded to 36.0% and 45.4%, yielding 26.0% and 24.0% biotin-GM2 as well as 10.0% and 21.4% biotin-GM3 after 72 hr and 120 hr at 37C, respectively. The metabolic products indicated that biotin-GM1 had reached degradative compartments of the cell and therefore could be used to study membrane flow along the endocytic pathway. In addition, 2.4% of biotin-GM1 was glycosylated to biotin-GD1a, very likely within the TGN.
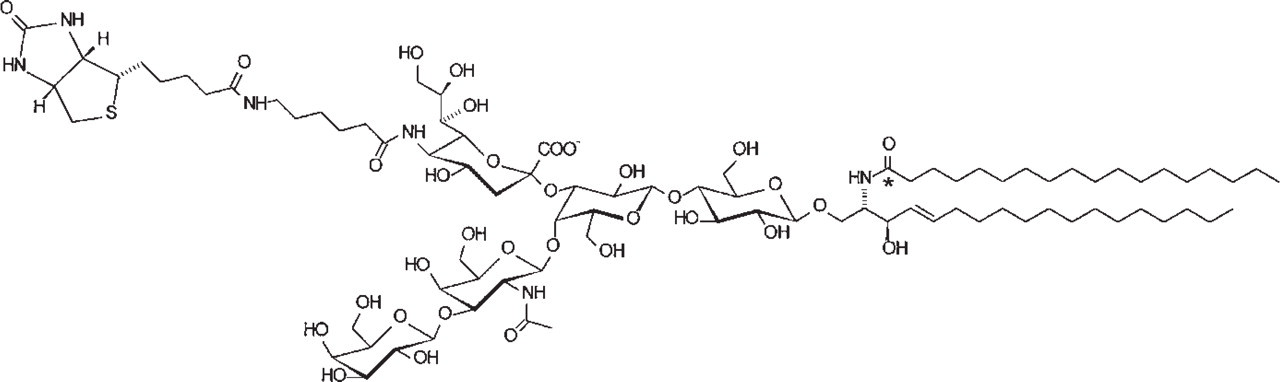
Structure of a biotin-labeled analogue of ganglioside GM1. Asterisk denotes position of radiocarbon.
Immunolabeling of LR Gold Sections
After 72 hr of incubation with biotin-GM1, mono-layer cells were harvested with proteinase K and formaldehyde-fixed. Cell pellets were postfixed with OsO4 followed by embedding in LR Gold, a plastic resin suitable for immunolabeling. Both the OsO4 fixation and the embedding at low temperature should minimize lipid redistribution. Quantification of the amount of radioactivity in the dehydration medium revealed that at most 10–15% of the cell-associated biotin-GM1 was extracted during dehydration. In comparison to this, about 70% of cell-associated biotin-GM1 was lost during conventional Epon embedding.
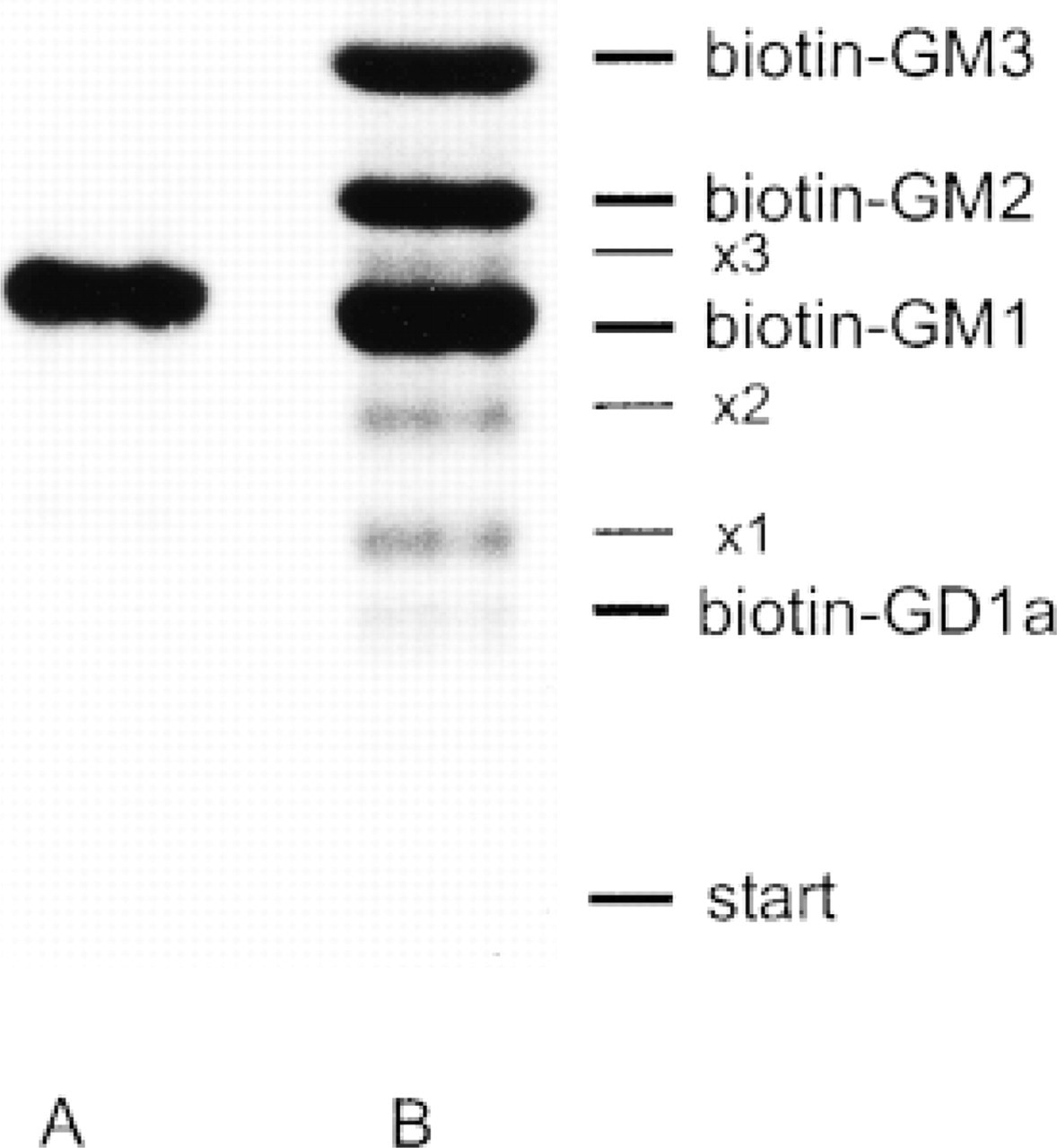
Metabolism of the biotin-labeled GM1 analogue in cultured human fibroblasts. Lane A, biotin-GM1 before incubation with cells. Lane B, lipid extract from cells incubated with biotin-GM1. The metabolic products are biotin-GM2, biotin-GM3, and biotin-GD1a as indicated. The identity of the radioactive bands was confirmed in previous studies (Albrecht et al. 1997). × 1, × 2, and × 3 indicate the sulfoxides of biotin-GM1, biotin-GM2, and biotin-GM3.
As shown in Figure 3, the biotin-GM1 is visible on the plasma membrane, in caveolae (Figure 3A), and in coated pits (Figure 3B). Moreover, the biotin-GM1 is detectable over the membranes of late endosomes and lysosomes, immunocytochemically identified by the use of antibodies specific for MPR (Figure 3C) or LAMP-1 (Figure 3D). Interestingly, the label for MPR and LAMP-1 is visible on the perimeter membrane, whereas the biotin-GM1 is mainly localized on the intraendosomal/intralysosomal membranes. Because of the OsO4 fixation, the membranes show an excellent contrast. The antibiotin label was always found close to membranes, suggesting minimal or no lipid redistribution during the dehydration steps in the embedding procedure. As expected, the labeling efficiency of the antibodies against MPR and LAMP-1 was quite low because these, like many other antigens, are sensitive to OsO4 fixation. When OsO4 fixation of the samples was omitted, the labeling efficiency was higher but membranes showed a poor contrast and details of organelle morphology could hardly be observed (Möbius 1998). To achieve optimal preservation of the ultra-structure combined with minimal interference with the preservation of antigens, we next employed immunolabeling of cryosections.
Immunolabeling of Cryosections
Fibroblasts were incubated with the biotin-GM1 for 72 hr as described and harvested with proteinase K. Fixed pellets were infused overnight with buffered 50% polyvinylpyrrolidone in 1.15 M sucrose as a cryoprotectant and frozen in liquid nitrogen. Frozen sections were picked up with a mixture of methylcellulose and sucrose according to Liou et al. (1996). Biotin-GM1 was distributed in patches over the plasma membrane (Figure 4A). As shown in Figure 4B, biotin-GM1 was detectable in caveolae, which were identified with anti-caveolin-1 antibodies. Furthermore, the biotin-GM1 was visible over the membranes of multilamellar organelles (Figures 4C and 4D). Only a few molecules of biotin-GM1 were detected on the internal vesicles of multivesicular bodies (Figures 4C and 4D). It appears that the biotin-GM1 and its metabolites are more concentrated in multilamellar than in multivesicular bodies, suggesting that these multilamellar organelles are lysosomes in which biotin-GM1 and its biotin-labeled metabolic products accumulate.
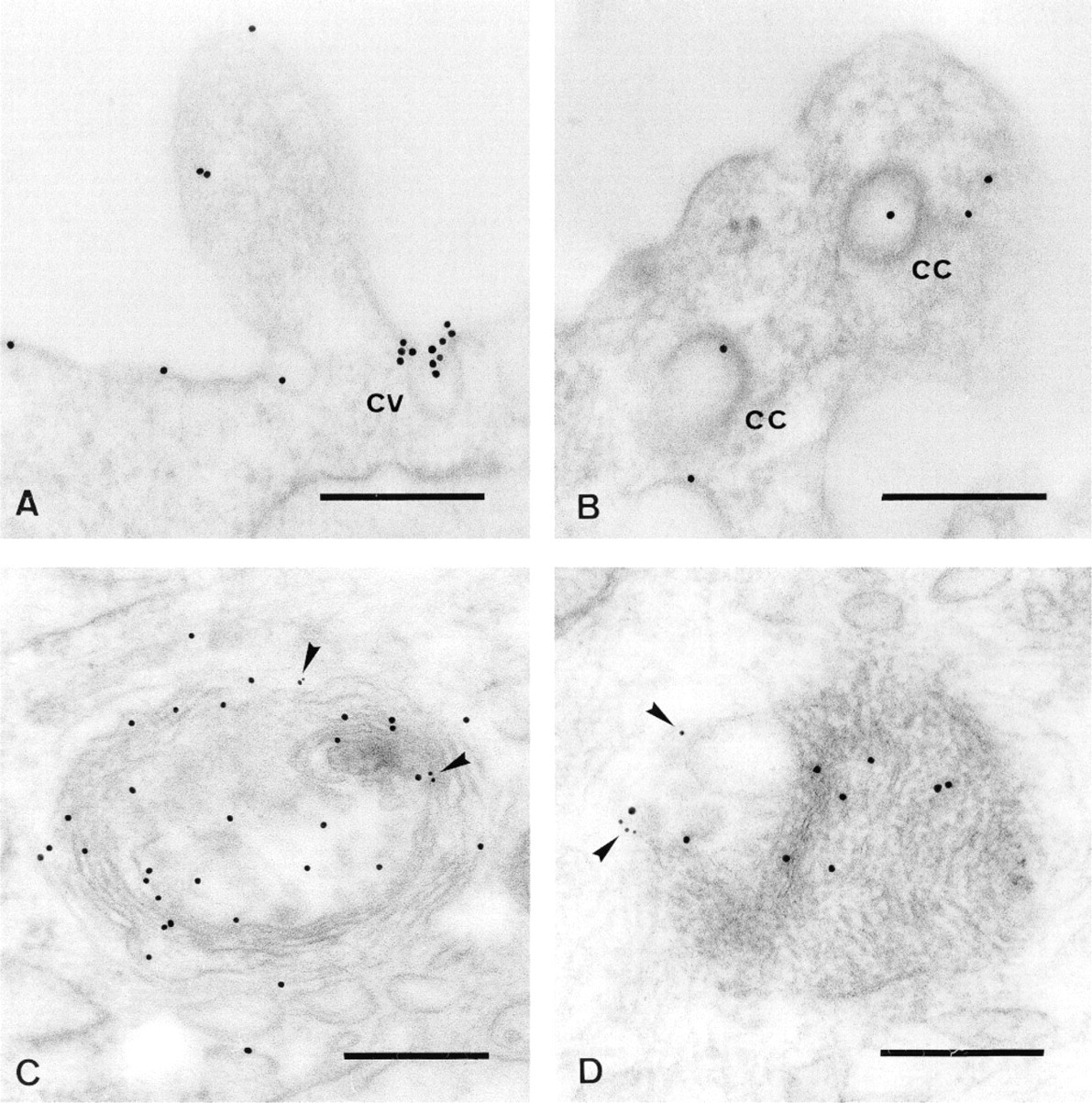
Immunolabeling of LR Gold sections. Human skin fibroblasts were incubated with biotin-GM1 for 72 hr as described. The GM1 analogue was detected with goat anti-biotin antibodies conjugated to gold (10 nm). The biotin-GM1 is concentrated in caveolae (cv) (A), but clathrin-coated pits or vesicles (cc) (B) were only occasionally labeled. The biotin-GM1 is also visible over the membranes of late endosomes and lysosomes, defined by labeling with antibodies against MPR (C, arrowheads) and antibodies against LAMP-1 (D, arrowheads). Bars = 0.2 μm.
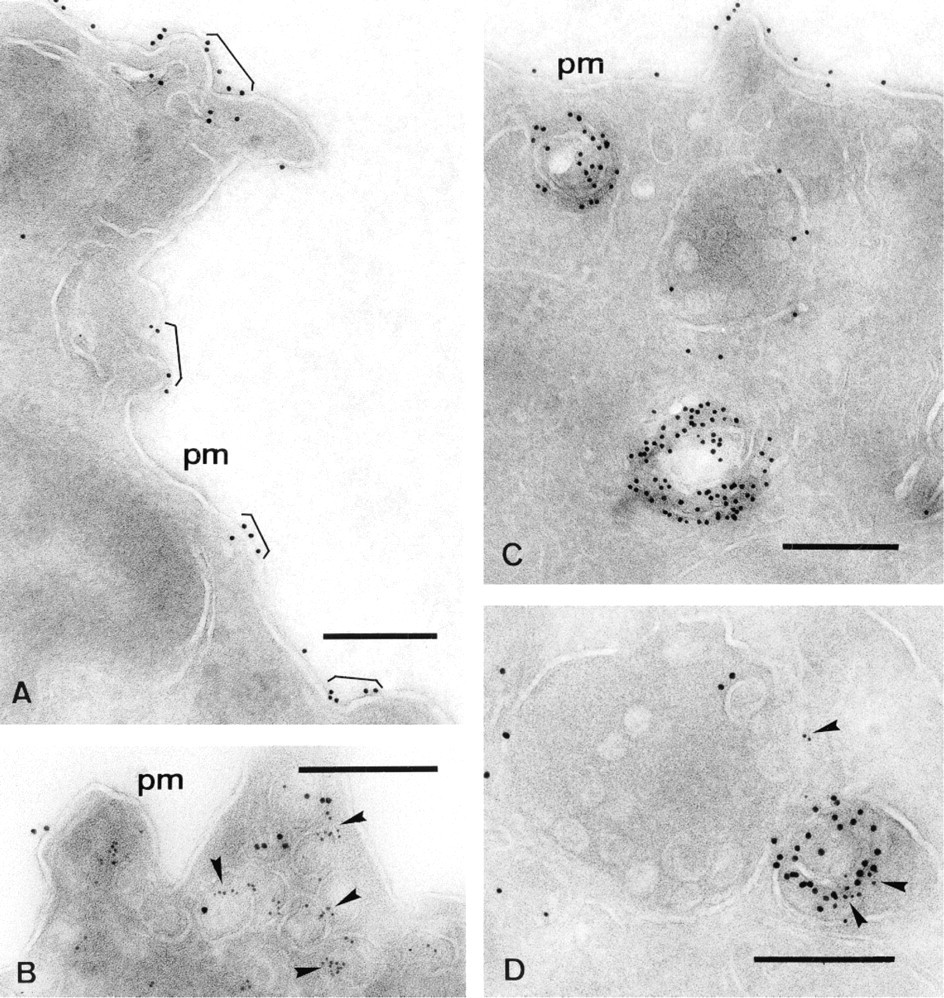
Immunolabeling of cryosections: Fibroblasts were incubated with biotin-GM1 for 72 hr as described. Cryosections were labeled with goat anti-biotin antibodies conjugated to gold (10 nm). The GM1 analogue is distributed over the plasma membrane (pm) in the form of patches (A) and is also detectable in caveolae, identified with anti-caveolin-1 antibodies (B, arrowheads), and over the membranes of multilamellar bodies (C,D). Double labeling with goat anti-biotin-gold (10 nm) and mouse anti-LIMP (D, 6-nm gold particles; arrowheads) shows that the biotin-labeled GM1 analogue is transported to the luminal membranes of multilamellar and multivesicular late endosomes or lysosomes. Bars = 0.2 μm.
Double labeling with goat anti-biotin and antibodies against LIMP (Figure 4D) revealed that the organelles are late endosomes and lysosomes. They contain many internal structures, such as vesicles and multilamellar membranes, that bear biotin-GM1. To find out whether these internal membranes are the place at which degradation of plasma membrane components, such as proteins and lipids, takes place, we analyzed the distribution of biotin-GM1 in comparison to the EGF receptor in EGF-stimulated fibroblasts.
Co-localization of Biotin-GM1 with the EGF Receptor in EGF-stimulated Fibroblasts by Immunoelectron Microscopy
The downregulation of the EGF receptor is a biochemically and morphologically well-characterized phenomenon and takes place in lysosomes (Haigler et al. 1979; Beguinot et al. 1984; Stocheck and Carpenter 1984; Hopkins et al. 1990; Renfrew and Hubbard 1991; Futter et al. 1996). We therefore used this system to find out whether the biotin-GM1 and the EGF receptor are sorted to the same intraendosomal/intralysosomal membranes for degradation.
For localization of the EGF receptor we used MAb (EGF-R1) that recognizes a determinant in the peptide portion of the extracellular domain of the EGF receptor without interfering with binding of EGF (Water-field et al. 1982; Beguinot et al. 1984). Because a long incubation period is required for insertion of an immunocytochemically detectable amount of exogenously supplied gangliosides, confluent fibroblasts were loaded with biotin-GM1 for 72 hr before stimulation with EGF for 45 min. We had established previously that the stimulated EGF receptor reaches lysosomes within 45 min (not shown).
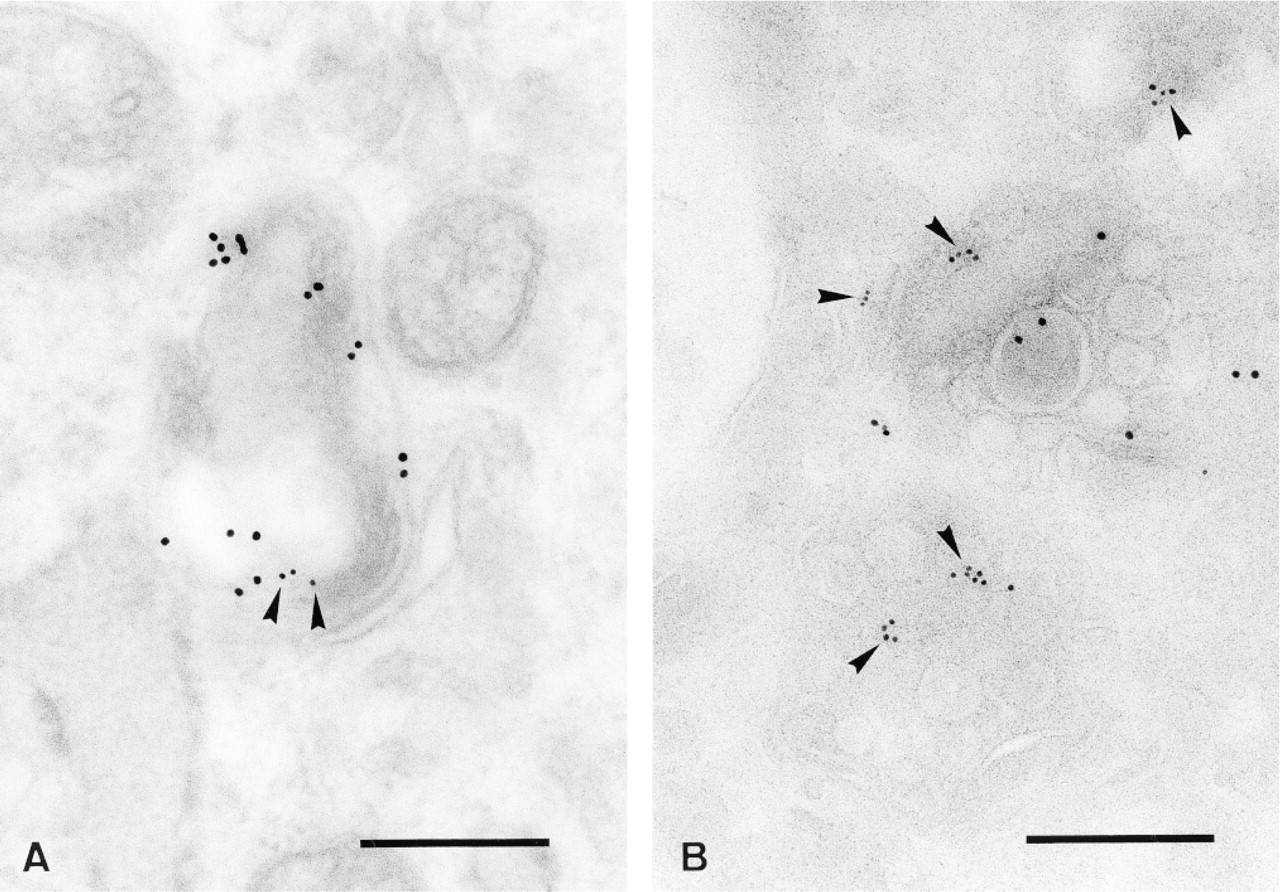
Localization of EGF receptor after 45-min incubation with EGF at 37C on LR Gold and cryosections. Fibroblasts were incubated with biotin-GM1 for 72 hr as described previously and were then incubated with EGF for 45 min at 37C. Sections were double labeled with goat anti-biotin antibodies conjugated to gold particles (10 nm) and mouse antibodies against the EGF receptor, detected with gold-conjugated goat anti-mouse antibodies (6 nm, arrowheads). On LR Gold sections (A) and cryosections (B) the biotin-labeled GM1 and the EGF receptor (arrowheads) are visible in the same compartment. Bars = 0.2 μm.
As shown in Figure 5, organelles are visible that are positive for both biotin-GM1 and EGF receptor on LR Gold sections as well as cryosections. The labeling efficiency of the anti-EGF-R1 antibody on LR Gold sections (Figure 5A) was low, probably due to loss of antigenicity during OsO4 fixation. The internal membranes are labeled with anti-biotin antibodies, indicating biotin-GM1, and the EGF receptor is detectable on the same structures. The cryosections show multivesicular bodies (Figure 5B) that bear label for biotin-GM1 and the EGF receptor on their internal vesicles. These results indicate that both the biotin-GM1 and the EGF receptor, after stimulation with EGF, were transported to internal membranes of late endosomes and lysosomes, indicating the same pathway of endocytosis.
Discussion
This article describes the localization of an exogenously added biotin-labeled analogue of GM1 along the endocytic pathway after insertion into the plasma membrane of cultured fibroblasts. This approach provides a number of advantages. It allows, for the first time, analysis of the fate of an individual labeled ganglioside by direct visualization. Moreover, it is possible to use labeled exogenous glycosphingolipids as membrane tracers in endocytosis, which is not possible with metabolically labeled endogenous gangliosides. For an exogenous glycosphingolipid to become a true tracer for membrane lipids in endocytosis, it must be inserted into and must become a component of the lipid layer of the plasma membrane.
Insertion of Biotin-GM1 into Cell Membranes
In aqueous solutions, gangliosides occur in the form of micelles, monomers, and oligomers in an equilibrium at concentrations above the CMC of about 10-9 M or less (Formisano et al. 1979; Mraz et al. 1980). Over many years, extensive studies on the binding to and uptake of gangliosides by biological membranes were performed (for an overview see Saqr et al. 1991). In principle, the binding of gangliosides to plasma membranes occurs in three different operationally determined fashions: loosely associated micelles removable by serum proteins, a protein-bound, serum-resistant but trypsin-labile ganglioside fraction, and trypsin-stable associated gangliosides largely comprising inserted monomers (Callies et al. 1977; Radsak et al. 1982). Treatment of cells with proteases such as trypsin removes the merely adsorbed ganglioside portion, leaving the truly incorporated ganglioside molecules with the cells. We have shown previously that, when applied under the same conditions, there was no substantial difference in the amount of cell-associated biotin-GM1 and GM1 in different cell types, indicating that the biotin label does not interfere with binding and insertion of labeled GM1 (Albrecht et al. 1997).
As demonstrated with spin-labeled ganglioside analogues (Schwarzmann et al. 1983, 1987), the trypsinstable portion represents incorporated ganglioside molecules. From the analysis of the electron spin resonance spectra, it could be shown that at least 70% of the incorporated spin-labeled gangliosides is intermixed with other lipids of the host membranes. The remainder of approximately 20% could represent either ganglioside molecules clustered in microdomains or ganglioside micelles endocytosed by the cells. Because the antibiotin-gold conjugate is always found associated with cell membranes, we are convinced that the detectable biotin tag represents gangliosides inserted into membranes.
The insertion into plasma membranes of gangliosides from their micellar state is a slow process. To incorporate significant amounts of the biotin-GM1 into cells, it was necessary to apply prolonged incubation times at 37C. We could show that maximal incorporation into cells of biotin-GM1 was reached after 72 hr. Further incubation did not result in increased incorporation.
Localization of Biotin-GM1 by Immunoelectron Microscopy
Studies of the intracellular distribution of lipids with the high resolution of electron microscopy require methods that maintain membrane structure and composition. Therefore, particular care must be taken to preserve membrane structure, especially in the case of lipid-rich membranes that are often poorly fixed. Previous studies had shown that the localization of Forssman glycolipid in cryosections was not satisfactory because of nonspecific labeling (van Genderen et al. 1991). This was probably caused by extraction and re-localization of the Forssman glycolipid during thawing. These artifacts were avoided by application of postembedding labeling techniques to sections of freeze-substituted cells. We could minimize lipid extraction using a protocol of progressive lowering of temperature and embedding in a hydrophilic resin. Best results were achieved using cryosections as a substrate for immunolabeling, as described by Liou et al. (1996), who found that the steps of thawing and transfer of the sections to the EM grid are most critical for the preservation of lipid-rich structures on cryosections. They significantly improved the structural preservation by substituting the conventional sucrose solution for section retrieval by a mixture of methylcellulose and sucrose (Liou et al. 1996). By applying their modified section retrieval protocol, we were able to localize bi-otin-GM1 over membranes of multilamellar organelles.
Endocytic Pathway of Biotin-GM1-labeled Membrane
Immunolabeling of LR Gold sections and cryosections revealed that the biotin-GM1 is detectable at the plasma membrane and that it occurs in the form of patches. In addition, it appears to be concentrated in caveolae, with only sparse labeling of clathrin-coated pits or vesicles. This result is in agreement with the findings of Parton (1994), who showed, using cholera toxin-gold conjugates, that endogenous GM1 can be found at the plasma membrane predominantly in caveolae, where GM1 is believed to be a part of sphingolipid-cholesterol microdomains or rafts (Fra et al. 1995; Harder and Simons 1997; Hooper, 1998). Therefore, biotin-GM1 appears to participate in the lateral segregation into rafts.
Moreover, biotin-GM1 could be detected intracellularly over intraendosomal and intralysosomal membranes. This clearly demonstrates that these internal membranes are in part plasma membrane-derived and reach the lumen of late endosomes and lysosomes during endocytosis. A similar targeting has been demonstrated for the EGF receptor after binding of EGF (Haigler et al. 1979; Felder et al. 1990; Hopkins et al. 1990; Futter et al. 1996). Therefore, we directly compared the localization of the biotin-GM1 analogue with that of the EGF receptor in fibroblasts after 45 min of incubation with EGF. We could demonstrate that biotin-GM1 and the EGF receptor co-localize on the internal membrane structures of the same organelle. This suggests that both the GM1 analogue and the EGF-EGF receptor complex follow the same pathway for degradation. This result supports the hypothesis that glycosphingolipids must become components of intraendosomal and intralysosomal vesicles before their degradation (Fürst and Sandhoff 1992; Sandhoff and Kolter 1996), as also suggested by the findings of Burkhardt and co-workers (1997). A similar topology of degradation has also previously been proposed for membrane proteins, such as growth factor receptors, in the process of their downregulation (Harding et al. 1985; Hopkins et al. 1990; Renfrew and Hubbard 1991; van Deurs et al. 1993).
The localization of biotin-GM1 on internal membranes of late endosomes and lysosomes is in accordance with the distribution of endogenous GM1 (Par-ton 1994). A similar distribution has been reported for the localization of the Forssman glycolipid in MDCK II cells by van Genderen and co-workers (1991), who showed an abundance of Forssman glycolipid in the internal membranes of multivesicular endosomes and lysosomes. The similarity in the intracellular distribution of biotin-GM1 with endogenous GM1 along the endocytic pathway, as described by Parton (1994) shows that biotin-GM1 behaves like endogenous GM1. Therefore, our observations demonstrate that biotin-GM1 is a powerful tool in studies of endocytosis of labeled membranes. It also provides insights into the localization and segregation properties of an individual ganglioside on cell membranes. Further investigation will clarify whether biotin-GM1 can be used as a marker for lipid microdomains. Moreover, we are now synthesizing a biotin-ganglioside derivative with a short acyl chain moiety in place of the natural long-chain residue. The short acyl chain analogues of lipids form 1:1 complexes with defatted BSA that allow insertion of these lipids into plasma membranes even under temperature block of endocytosis (Pagano 1989; Schwarzmann et al. 1995). This offers the possibility of pulse-chase experiments to investigate time-dependent events in the behavior of an individual ganglioside during endocytosis.
To further investigate the properties and roles of individual glycosphingolipids in intracellular membrane transport, the synthesis of other biotin-labeled glycosphingolipid derivatives and the immunolocalization after their uptake by cells are of major interest.
Footnotes
Acknowledgments
Supported by a grant from the Deutsche Forschungsgemeinschaft (SFB 284/B5 to GS).
We are grateful to Dr Bernd Hoflack, Institut Pasteur, Lille, France, for the generous gift of rabbit anti-MPR antibodies. WM wishes to thank Drs Gareth Griffiths and Paul Webster for helpful discussions during the EMBO course in Prague, 1997.