
Abstract
In situ hybridization (ISH) using nonradioactive probes enables mRNAs to be detected with improved cell resolution but compromised sensitivity compared to ISH with radiolabeled probes. To detect rare mRNAs, we optimized several parameters for ISH using digoxygenin (DIG)-labeled probes, and adapted tyramide signal amplification (TSA) in combination with alkaline phosphatase (AP)-based visualization. This method, which we term TSA-AP, achieves the high sensitivity normally associated with radioactive probes but with the cell resolution of chromogenic ISH. Unlike published protocols, long RNA probes (up to 2.61 KB) readily permeated cryosections and yielded stronger hybridization signals than hydrolyzed probes of equivalent complexity. RNase digestion after hybridization was unnecessary and led to a substantial loss of signal intensity without significantly reducing nonspecific background. Probe concentration was also a key parameter for improving signal-to-noise ratio in ISH. Using these optimized methods on rat taste tissue, we detected mRNA for mGluR4, a receptor, and transducin, a G-protein, both of which are expressed at very low abundance and are believed to be involved in chemosensory transduction. Because the effect of the tested parameters was similar for ISH on sections of brain and tongue, we believe that these methodological improvements for detecting rare mRNAs may be broadly applicable to other tissues.
I
Catalyzed reporter deposition (CARD) is a recently developed method (Bobrow et al. 1991,1992) that might potentially address the problem of detecting low-copy mRNAs by ISH. The method uses horseradish peroxidase to catalyze the deposition of tyramide molecules, preconjugated with either biotin or fluorescent reporters. The method, also called tyramide signal amplification (TSA), has been adopted as a means of enhancing signal strength for immunoblotting and immunocytochemistry (Bobrow et al. 1991,1992; Chao et al. 1996; van Gijlswijk et al. 1997) and fluorescent in situ hybridization (FISH) on chromosomes (Kerstens et al. 1995; Raap et al. 1995; Speel et al. 1997). More recently, TSA has also been adapted for ISH detection of cellular mRNAs (Schmidt et al. 1997; Wanner et al. 1997). Using biotinyl-tyramide and streptavidin conjugated to alkaline phosphatase (AP) to introduce an additional round of amplification, we have now achieved a considerable enhancement of signal intensity relative to standard chromogenic ISH detection systems. We term this protocol TSA-AP (tyramide signal amplification using AP). In addition, we have optimized critical parameters to improve hybridization with digoxigenin (DIG)-labeled probes and to reduce nonspecific binding of probes, both strategies being essential for successful detection of rare mRNAs.
We have been studying rare mRNAs in rat taste buds, the sensory receptor organs for gustation. By improving signal-to-noise ratio in ISH, we are now able to detect mRNAs in taste bud cells for gustducin, a taste-specific G-protein, (McLaughlin et al. 1992), for transducin (Tanabe et al. 1985), the phototransduction G-protein that is closely related to gustducin, and for a metabotropic glutamate receptor, mGluR4 (Tanabe et al. 1992). Gustducin and transducin are believed to be involved in bitter and sweet transduction (Wong et al. 1996; Ming et al. 1998). mGluR4 is a G-protein-coupled glutamate receptor found in taste buds and postulated to be involved in taste transduction for monosodium glutamate (Chaudhari et al. 1996; Bigiani et al. 1997).
Materials and Methods
Reagents
Transcription reagents, including DIG-11-UTP and T7 RNA polymerase, anti-DIG-Fab conjugated with either horseradish peroxidase (HRP) or alkaline phosphatase (AP), streptavidin conjugated with alkaline phosphatase, 4-nitroblue tetrazolium chloride (NBT), and 5-bromo-4-chloro-3-indolyl-phosphate (BCIP), were purchased from Boehringer Mannheim Biochemical (Indianapolis, IN). Biotinyl-tyramide was purchased from New England Nuclear (Boston, MA) as a component of their Renaissance TSA-Indirect system. Formamide (molecular biology grade, VWR) was not further deionized before use. Bovine serum albumin (BSA), polyvinylpyrrolidone, Ficoll, yeast tRNA, salmon sperm DNA, and levamisole were purchased as molecular biology grade (RNase-free) reagents from Sigma (St Louis, MO). Dextran sulfate (MW 500,000; molecular biology grade) was from Pharmacia (Uppsala, Sweden). Proteinase K, RNase A, and most other general molecular biology reagents were purchased from Bethesda Research Laboratories (Gaithersburg, MD).
Solutions
All buffers and salt solutions before the detection steps were sterile-filtered, treated with 0.1% diethyl pyrocarbonate (DEPC), and autoclaved. Solutions containing proteins or other macromolecules were prepared with RNase-free components dissolved in water pretreated with DEPC. The following solutions were used:
Phosphate-buffered saline (PBS): 145 mM NaCl, 1.4 mM KH2PO4, 8 mM Na2HPO4 (pH 7.4).
20 x SSC: 3 M NaCl, 300 mM sodium citrate (pH 7.0 adjusted with citric acid).
50 x Denhardt's: 1% bovine serum albumin, 1% polyvinylpyrrolidone, 1% Ficoll.
Hybridization buffer: 50% formamide, 2 x SSC, 1 x Denhardt's, 10% dextran sulfate, 0.5 mg/ml yeast tRNA, and 0.5 mg/ml salmon sperm DNA (hydrolyzed according to Wisden et al. 1991). One-ml aliquots were stored at −20C.
STE: 500 mM NaCl, 20 mM Tris-HCl (pH 7.5), 1 mM EDTA.
Wash buffer: 100 mM maleic acid, 150 mM NaCl, 0.3% Tween-20 (pH 7.5).
Blocking buffer 1: 1% blocking reagent (Boehringer Mannheim) in wash buffer.
Blocking buffer 2: 0.5 % blocking reagent (Renaissance; NEN) in 150 mM NaCl, 100 mM Tris-HCl (pH 7.5).
Detection buffer: 100 mM NaCl, 100 mM Tris-HCl (pH 9.5), 50 mM MgCl2.
Substrate solution: 337 μg/ml NBT, 175 μg/ml BCIP, 5 mM levamisole in detection buffer.
TE: 10 mM Tris-HCl (pH 7.5), 1 mM EDTA.
cDNA Clones and RNA Probes
All cDNA inserts were generated by Reverse Transcriptase-polymerase chain reaction (RT-PCR) using poly(A)RNA from rat brain, retina, or tongue as template. RT-PCR products were blunt-cloned into the EcoRV site of the Bluescript vector, pKS-, in both orientations. The short probe for mGluR4 (0.8 KB) was transcribed from a previously described clone spanning amino acids 409–676 (Chaudhari et al. 1996). For the long mGluR4 probe (2.6 KB), the cDNA insert begins at amino acid 291 and continues through nucleotide 695 of the 3′ untranslated region of rat mGluR4a (Tanabe et al. 1992). A 1.03-KB insert for rat α-rod transducin was generated by RT-PCR, using rat retina cDNA as template, and corresponding to amino acids 6–347 of the bovine rod transducin sequence (Tanabe et al. 1985). A short (0.27 KB) clone for rat gustducin insert, spanning amino acids 48–136, was provided by Dr. R. Margolskee. A longer insert (1.37 KB) for rat gustducin was generated by RT-PCR, using cDNA from rat lingual tissue mRNA as template, and included the entire coding sequence as well as 83 BP of 5′- and 232 BP of 3′ untranslated sequences, respectively (McLaughlin et al. 1992). The DNA sequence identity between our gustducin and transducin probes is 71% in the region of overlap (1.03 KB).
Digoxigenin-labeled RNA (DIG-RNA) probes were transcribed in anti-sense and sense orientation using T7 RNA polymerase. We have previously reported that probes transcribed from pKS using T3 RNA polymerase tend to yield higher background binding in lingual epithelium and therefore are less suitable as ISH probes for certain tissues (Chaudhari et al. 1996). Some probes were fragmented to determine the effect of probe size on hybridization efficacy (see Results). Limited alkaline hydrolysis was performed by incubating the RNA probe in 60 mM Na2CO3, 40 mM NaHCO3, at 60C for a predetermined time that depended on starting RNA length and a desired final length of 0.3 KB (Angerer and Angerer 1992). Only RNAs that migrated as sharp bands (such as those shown in Figure 6E) of the correct molecular length in agarose gel electrophoresis were used as in situ hybridization probes. To estimate probe concentration, serial dilutions of labeled probes and a standard labeled RNA were spotted on nylon membranes (Hybond N+; Amersham, Arlington Heights, IL), immunodetected using 1:5000 diluted anti-DIG-Fab-AP conjugate, and visualized with NBT and BCIP according to instructions from Boehringer Mannheim. Probes were diluted with DEPC-treated water to 5 ng/μl and were stored in aliquots at −80C.
Preparation of Tissue and Sectioning
Tongues or brains were dissected rapidly from preweaning (for mGluR4) or adult (for all other probes) Sprague-Dawley rats after sacrifice by CO2. In rat, a single vallate and two foliate papillae, found on the dorsal surface of the tongue, contain large numbers of taste buds. For preparing cryosections, fresh tissues were placed in chilled OCT and snap-frozen in isopentane precooled in dry ice. Tissue blocks were stored at −80C for up to 4 weeks. Cryosections of 10–12 μm were cut at −20C, collected on baked Superfrost Plus slides (Fisher Scientific; Fair Lawn, NJ), and stored desiccated at −80C. For detection of rare mRNAs in taste buds, we used cryosections stored for no longer than 2 days.
Fixation and Pretreatment of Sections
Fixation and all pretreatment steps were carried out in baked glass Coplin jars on a gyrotary shaker with gentle agitation. A key role of fixation is to maximize mRNA preservation. Therefore, cryosections were removed from −80C storage and were immediately fixed in freshly prepared 4% paraformaldehyde in PBS at 4C for 20 min. We found that prolonged thawing of unfixed sections or fixation at room temperature (RT) reduced signal intensity. Alternatively, tissues were immersion-fixed before cryosectioning, as previously described (Chaudhari et al. 1996).
Sections were rinsed twice in PBS for 5 min each. Endogenous peroxidase activity was quenched with 1% H2O2 in methanol for 15 min, followed by two rinses in PBS of 5 min each. Endogenous AP activity was quenched with 0.2 M HCl for 8 min, followed by two 5-min washes in PBS. For tissue permeabilization, the optimal concentration of proteinase K during digestion of lingual and brain sections was determined empirically (not shown). For partial proteolysis, sections were permeabilized with 10 μg/ml proteinase K in 10 mM Tris-HCl (pH 7.5) at RT for 5 min (brain) or 10 min (lingual tissue). Detergent treatment was 1% Triton X-100 in PBS for 10 min. After rinsing in 10 mM Tris-HCl, sections were equilibrated in 0.1 M triethanolamine (TEA) for 2 min and then were acetylated in freshly prepared 0.25% acetic anhydride in 0.1 M TEA for 10 min with vigorous shaking. After rinsing in 2 x SSC for 10 min at 45C, sections were air-dried for 5 min on a slide warmer at 60C. Sections were encircled with rubber cement to enclose hybridization buffer and were used immediately in hybridization.
Hybridization
DIG-RNA probes were freshly diluted in 1 ml hybridization buffer and denatured at 80C for 5 min. Hot probe in hybridization buffer (50 μl/section) was directly applied to dry preheated sections. Slides were incubated overnight in a humid chamber containing paper towels moistened with 50% formamide and 2 x SSC. The temperature for hybridization and posthybridization washes was adjusted according to the GC content of each probe and was selected for high stringency: 45C for gustducin (GC content 41%), 50C for transducin (GC content 52%), and 55−60C for mGluR4 (GC content 60%).
Posthybridization Washes
Sections were washed in 2 x SSC at 45C for 10 min to remove excess probe. For slides treated with RNase (see Results), sections were equilibrated in STE, incubated with 10 μg/ml RNase A in STE for 30 min at RT, and washed for 10 min at 50C in STE. Sections were then subjected to three high-stringency washes: twice in 50% formamide, 1 x SSC at the hybridization temperature for 20 min each time, followed by a 20-min wash in 0.2 x SSC at the same temperature.
Immunodetection
The reagents and conditions in this section are essentially as recommended by Boehringer Mannheim. Slides were rinsed in wash buffer at RT. Nonspecific binding was blocked in freshly prepared blocking buffer 1 for 30 min at 37C. Sections were incubated for 1 hr at RT with anti-DIG-Fab-AP conjugate (diluted 1:500) or anti-DIG-Fab-HRP (diluted 1:100, for slides used in TSA-AP) in blocking buffer 1. Sections were washed three times for 5 min each in wash buffer. For slides subjected to TSA-AP, sections were blocked again for 30 min at 37C in blocking buffer 2. Biotinyl-tyramide (BT) was diluted (1:50-1:100 in the diluent provided) and sections were incubated in the dark for no longer than 10 min at RT in this substrate. After three washes of 5 min each in wash buffer, streptavidin-AP (diluted 1:750 in blocking buffer 1) was applied to the sections for 1 hr at RT in the dark. Finally, sections were rinsed three times for 5 min each in wash buffer.
For unamplified (i.e., AP protocol) and TSA-AP protocols alike, sections were first equilibrated in detection buffer. Sections were incubated in substrate solution in a humid chamber at RT in the dark. The reaction was continued for up to 96 hr for the AP protocol or up to 4 hr for TSA-AP, then was stopped by rinsing slides in TE followed by water. Sections were mounted in Gelmount.
Vibratome Sections
To prepare vibratome sections, blocks of lingual tissue containing vallate or foliate papilla were immersion-fixed for 3 hr in fresh 4% paraformaldehyde in PBS at RT. Thick sections (40 μm) were cut in ice-cold PBS using a vibratome. Sections were used immediately and were maintained free-floating through solution changes in sterile multiwell culture dishes. A fine brush, treated with RNase Away (BRL) was used to manipulate sections. Vibratome sections were post-fixed in cold 4% paraformaldehyde in PBS for 15 min. With the following exceptions, reagents and procedures were as described for cryosections. After washing and pretreatment steps, sections were prehybridized for 30 min at 60C in hybridization buffer lacking probe. The blocking steps for immunodetection were prolonged to 1 hr each. After completion of the color reaction, vibratome sections were transferred to 0.5% gelatin, spread on glass slides, allowed to dry, and mounted with Gelmount.
Northern Blots
In vitro transcribed sense RNA for gustducin and transducin was electrophoresed in a denaturing agarose gel and blotted as previously described (Chaudhari and Beam 1993). Blots were hybridized in the same buffer as for ISH (with dextran omitted) and using the same DIG-labeled RNA probes as used for ISH. Hybrids were immunodetected using anti-DIG-Fab-AP conjugate (diluted 1:5000), followed by a chemiluminescent substrate, CDP-Star (Amersham Life Science) and were visualized on X-ray film. Band intensities were quantified using a CCD camera and digitizing software (Alpha Innotech; San Leandro, CA).
Results
This study was initiated to use ISH to detect mRNAs for proteins believed to be involved in signal transduction in gustatory receptor cells. Such mRNAs include the taste-specific G-protein gustducin, the closely related G-protein transducin, and the metabotropic glutamate receptor mGluR4. Gustducin mRNA is known to be abundant, whereas the mRNAs for transducin and mGluR4 are found at low copy numbers in taste buds (McLaughlin et al. 1993; Ruiz-Avila et al. 1995; Chaudhari et al. 1996). To achieve the high sensitivity required for low-abundance mRNAs, we optimized crucial steps of the hybridization and detection to achieve the best possible signal-to-noise ratios. Because the effect of the tested parameters was similar for ISH on sections of brain and tongue, we believe that these methodological improvements for detecting rare mRNAs may be broadly applicable to other tissues. For mGluR4 probe, optimization experiments were conducted on sections of cerebellum in which the mRNA concentration is moderate. This allowed clear visualization of the effect of each change in ISH parameter.
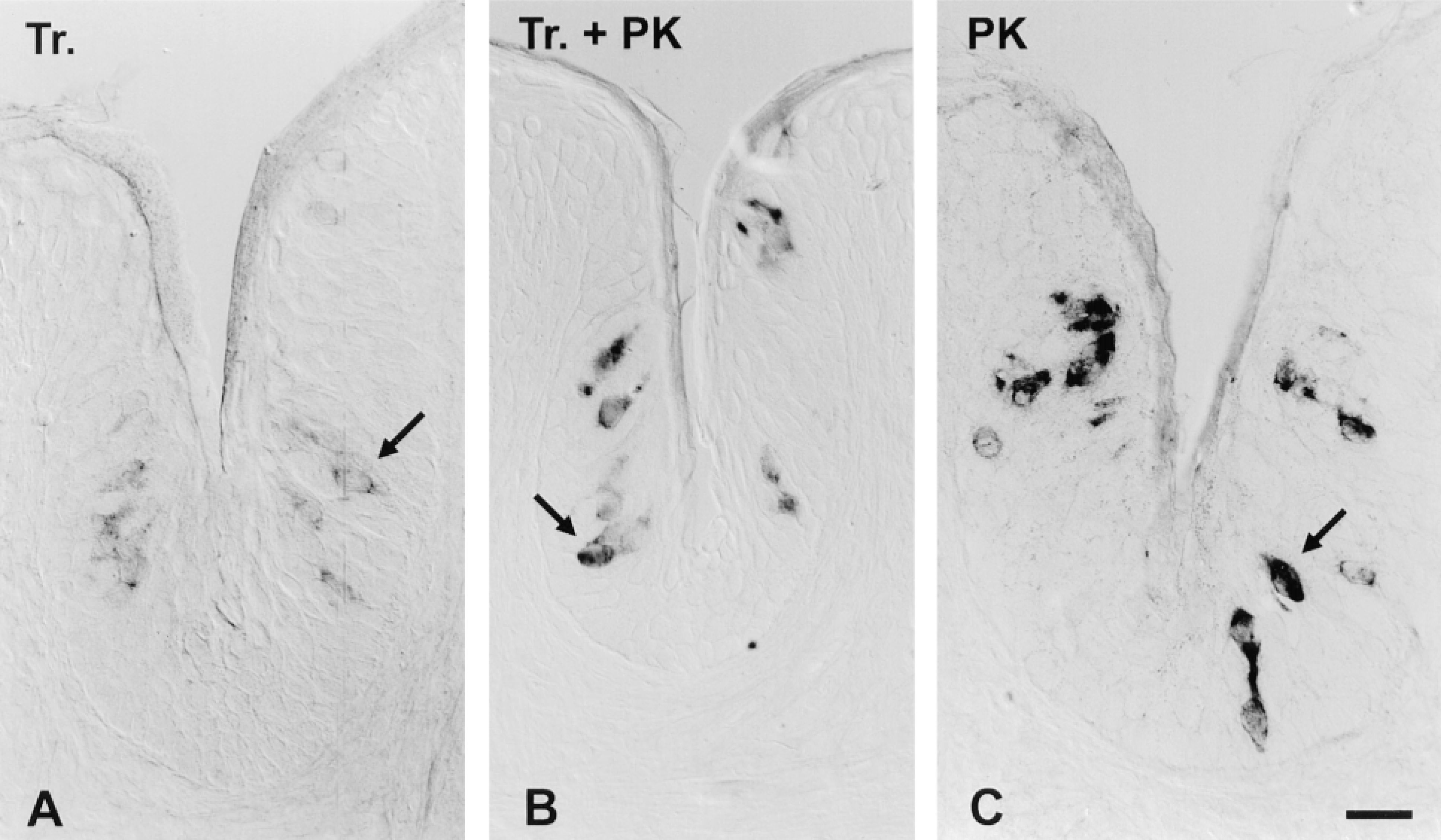
Effect of tissue permeabilization treatments on ISH signal intensity. Cryosections of foliate papilla, containing taste buds, were pretreated with (A) 1% Triton (Tr) for 10 min; (B) 10 μg/ml proteinase K for 10 min followed by 1% Triton (Tr + PK) for 10 min; or (C) 10 μg/ml proteinase K (PK) for 10 min. After pretreatments, all sections were hybridized in parallel at high stringency with a 1.37-KB anti-sense DIG probe for gustducin. No RNase treatment was employed and duplexes were visualized using the unamplified AP protocol with anti-DIG-AP and substrates NBT and BCIP. Hybridization signal (dark color) is limited to taste bud cells (arrow) and is missing from surrounding nonsensory epithelium and other cell types. Color development was allowed to proceed for 2.5 hr for each section. Bar = 20 μm.
Optimizing Hybridization and Detection
Tissue Permeabilization. The ability of ISH probes to permeate sections has a critical effect on the strength of the hybridization signal and must be optimized for each tissue of interest. We compared signal intensity with Triton and proteinase K treatments (Singer et al. 1986; Schaeren-Wiemers and Gerfin-Moser 1993; Panoskaltsis-Mortari and Bucy 1995) on lingual tissues. When a gustducin probe was used, detergent-treated sections showed weak staining of taste bud cells (Figure 1A) whereas proteinase K digestion resulted in a stronger signal (Figure 1C). The combination of both treatments, detergent and protease, yielded a weaker signal than proteinase K digestion alone (Figure 1B), implying that Triton treatment may have a deleterious effect on the target mRNA in cells. Proteinase K treatment also increased the hybridization efficiency on cerebellar sections using a probe for mGluR4 (not shown).
Detection Protocol with Tyramide Signal Amplification. Conventional chromogenic detection using the AP protocol yielded insufficient sensitivity to detect low-abundance mRNAs. The TSA Indirect system (NEN) allows signal amplification by using HRP (linked to anti-DIG-Fab) to deposit BT at the site of the DIG-labeled hybrids. We have combined this with a second amplification step using streptavidin-AP. The resulting protocol, termed TSA-AP, strongly enhanced specific hybridization signals compared to conventional chromogenic signal detection. Figures 2A and 2B compare signal intensity for mGluR4 probe in rat cerebellar sections, detected with the unamplified AP protocol or with TSA-AP. The ISH signal is localized to granule cells, as previously documented (Tanabe et al. 1993; Ohishi et al. 1995). The intensity of the hybridization signal was markedly stronger after TSA-AP visualization. We also applied this amplification protocol to lingual tissue, using a gustducin probe. Again, a definite increase in the intensity of the specific hybridization signal in taste receptor cells was seen (Figures 2C and 2D). In initial experiments, TSA-AP produced relatively high nonspecific background staining, seen as many dense purple particulates scattered throughout the tissue (Figure 2D). Although this might be acceptable for a moderately abundant mRNA (such as gustducin), detecting rare mRNAs necessitates a very clean background (Chaudhari et al. 1996).
Probe Concentration. In an attempt to reduce nonspecific deposits and background staining with TSA-AP, we systematically varied probe concentration during hybridizations. Hybridizing lingual sections with gustducin probes at 300 ng/ml resulted in high nonspecific staining with sense and anti-sense probe alike (Figures 3A and 3B). In particular, globular precipitates not associated with any apparent biological structures appeared throughout the tissue sections for probe in both orientations. Such nonspecific precipitates were eliminated after a tenfold reduction in probe concentration during hybridization (Figure 3C). Nevertheless, specific hybridization signal remained equally intense at the lower probe concentration (Figures 3B and 3C). This result implies that signal intensity is limited by the abundance of the target mRNA rather than by probe concentration, whereas nonspecific noise appears to be determined in part by probe concentration. We have also observed this striking improvement in signal-to-noise ratio after probe dilution when TSA-AP was employed for other genes (not shown). Therefore, optimal probe concentrations for TSA-AP are tenfold lower than those recommended (100–500 ng/ml) in many protocols for ISH with unamplified chromogenic detection (Schaeren-Wiemers and Gerfin-Moser 1993).
RNase Digestion. Many published ISH protocols call for treating sections with RNase after hybridization to improve specificity (Wahle and Beckh 1992; Litman et al. 1993; Panoskaltsis-Mortari and Bucy 1995). To test this, cerebellar sections were hybridized with mGluR4 sense and anti-sense probes. Posthybridization washes included or omitted a digestion step with 10 μg/ml RNase A. Sections not treated with RNase demonstrated considerably stronger hybridization signal for mGluR4 in the granule cell layer compared to sections digested with RNase (Figures 4A and 4B). Importantly, omitting RNase did not increase nonspecific staining, e.g., in the white matter or in the molecular layer of the cerebellum (Figure 4C). The faint nonspecific staining in Purkinje cells was visible in sections whether RNase digestion was included or omitted. Similar results were also obtained using lingual sections hybridized with gustducin probes (not shown).
Probe Length and Complexity. Complexity refers to the length of unique sequence in a probe. Increased probe complexity allows hybridization over a greater length of each target mRNA, thereby increasing the number of DIG moieties that label the target. This leads to increased signal intensity. Many published ISH protocols recommend hydrolyzing long RNA probes to shorter fragments, 150–300 bases in length, to improve their permeation into tissue sections while retaining complexity. We have examined the effect of both the physical length and the complexity of probes on hybridization efficacy. We systematically varied these independent parameters for mGluR4 and gustducin probes.
For each gene, we transcribed RNA probes from two separate clones that contained short or long inserts. For mGluR4, the cDNAs were either 0.8 or 2.6 KB long, whereas for gustducin, cDNA inserts were either 0.27 or 1.3 KB long. The hybridization signal for mGluR4 on cerebellar granule cells was considerably stronger with the 2.6-KB probe compared to the 0.8-KB probe (Figures 5A and 5C). Similarly, the hybridization signal for gustducin was considerably stronger with the 1.3-KB probe relative to the 0.3-KB probe (Figures 5D and 5F). Background staining with sense probes of each length was very low in all cases and was independent of probe complexity (not shown).
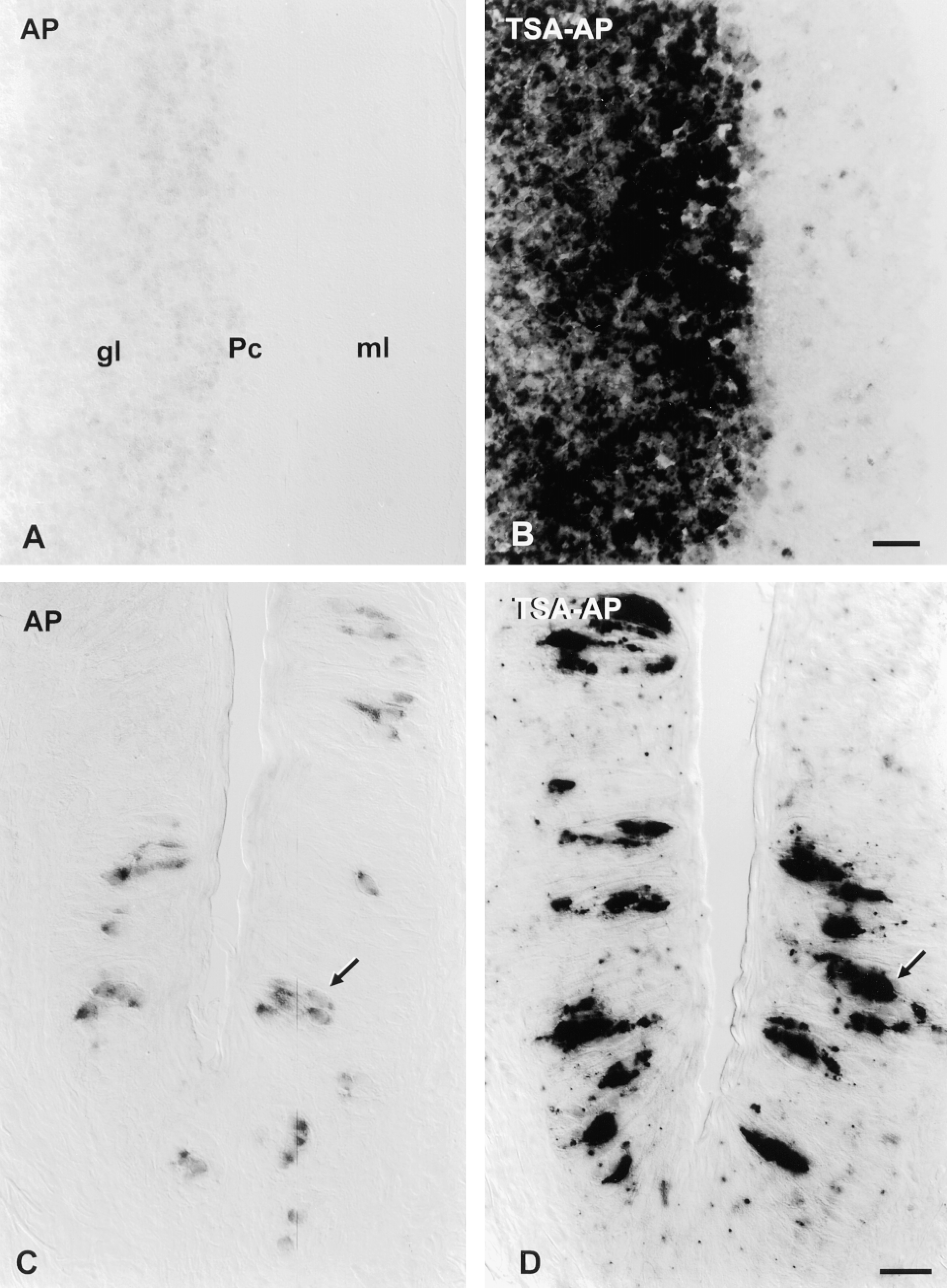
TSA-AP protocol increases hybridization signal intensity relative to unamplified AP protocol. Cryosections of cerebellum (A,B) were hybridized in parallel with a 2.6-KB anti-sense DIG probe for mGluR4; cryosections of vallate papilla (C,D) were hybridized in parallel with a 0.27-KB anti-sense DIG probe for gustducin. In each case, sections were permeabilized with proteinase K; no RNase digestion was employed after hybridization. Signal was visualized using either the AP protocol (A,C) or the TSA-AP protocol (B,D). For mGluR4, color development proceeded for either 4.5 days (unamplified protocol, A) or 1 hr (TSA-AP protocol, B). Bar = 40 μm. For gustducin probes, the color reaction was for either 18 hr (unamplified, C) or 30 min (TSA-AP, D). Bar = 20 μm. In the cerebellum, hybridization signal is limited to the granule cell layer (gl) and is very low in the molecular layer (ml), as expected. In lingual tissue, some taste bud cells (arrows) show signal. In each tissue, the color intensity of hybridization signal is considerably darker when TSA-AP is used.
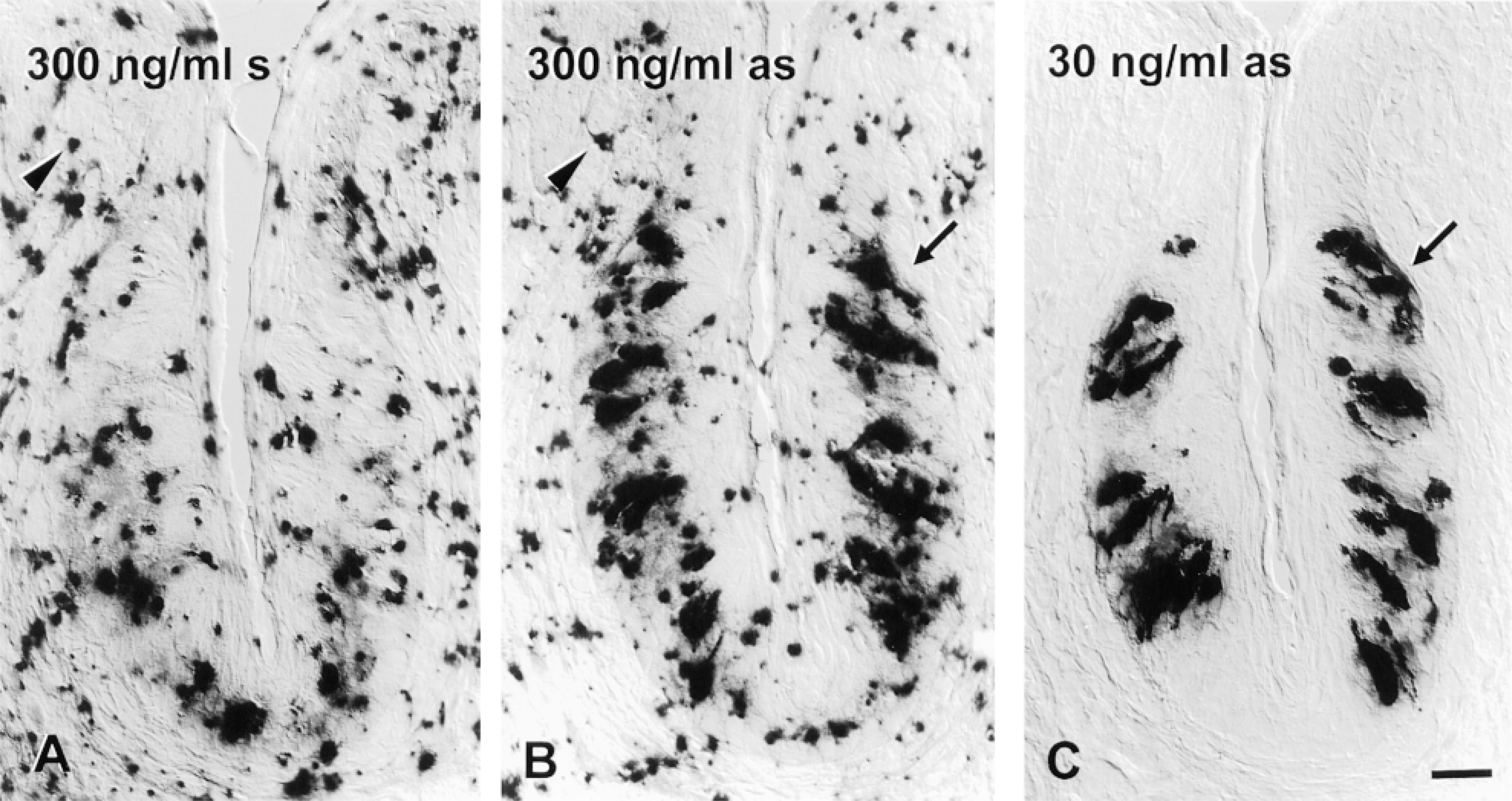
Probe concentration during hybridization determines signal-to-noise ratio in TSA-AP detection. Cryosections of foliate papilla were pretreated (including proteinase K digestion), hybridized with 1.37-KB gustducin probes, and detected in parallel using the TSA-AP protocol. Hybridization buffer contained (A), 300 ng/ml DIG-sense (s) RNA, (B), 300 ng/ml DIG anti-sense (as) RNA, or (C) 30 ng/ml DIG anti-sense RNA. In each case, the color development was allowed to proceed for 40 min. Particulate deposits (arrowheads), which constitute nonspecific background, are visible in both sections hybridized at high probe concentration (A,B) but are almost completely missing from sections hybridized at lower probe concentration (C). Signal intensity in taste bud cells (arrows) appears equally dark at both concentrations of probe. Bar = 20 μm.
We also tested the premise that short probes permeate into sections more effectively. Unexpectedly, signal intensity with the intact 2.6-KB mGluR4 probe was much stronger than when the same probe was reduced to 0.3-KB fragments by alkaline hydrolysis (Figures 5B and 5C). The same result was evident with an intact 1.37-KB gustducin probe compared to its hydrolyzed, shortened form (Figures 5E and 5F). Contrary to published protocols, our results show that fragmentation of long probes is an unnecessary step for ISH on cryosections and may even be detrimental. Proteinase K digestion before hybridization appears to allow RNA probes several kilobases long to penetrate 10-μm cryostat sections (and 40-μm vibratome sections; see below). Presumably, increased physical length of probe leads to more stable hybrids that survive stringency washes more effectively.
Applying TSA-AP to Detect Rare mRNAs in Taste Cells
We have used the enhancements described above (Table 1) to examine the expression of mRNAs for two G-proteins and a G-protein-coupled seven-transmembrane receptor in taste bud cells. Gustducin and transducin are closely related members of the G-protein superfamily. Gustducin mRNA is strongly expressed in taste receptor cells (McLaughlin et al. 1992), whereas mRNAs for transducin and mGluR4 are much less abundant in lingual tissue (McLaughlin et al. 1993; Chaudhari et al. 1996).
Transducin mRNA Is Expressed in a Small Fraction of Taste Bud Cells. Transducin is the principal G-protein involved in phototransduction. Transducin mRNA is detectable in lingual epithelia by RT-PCR and RNase protection (McLaughlin et al. 1993). Attempts to localize transducin mRNA to taste buds by ISH using radiolabeled probes met with limited success, probably because the low abundance of the mRNA did not produce a strong signal relative to nonspecific background (McLaughlin et al. 1993). Therefore, we applied TSA-AP to determine whether transducin could be detected more definitively in taste bud cells.
Figure 6A shows staining of taste receptor cells in a section of foliate papilla hybridized with a transducin anti-sense probe. An adjacent section, hybridized with transducin sense probe, does not show any signal (Figure 6B). Similar results were obtained in circumvallate papillae. To visualize hybridization with transducin probe in taste buds, it was necessary to use the TSA-AP protocol and longer reaction times compared to hybridization with gustducin probe. Signal was apparent within 5 min of color development for gustducin, in contrast to 15–60 min necessary for transducin. This difference in color reaction time corresponds to the higher level of expression for gustducin relative to transducin mRNA in lingual tissue, shown by RNase protection assay (McLaughlin et al. 1993). As expected, the transducin anti-sense probe gave a robust signal in photoreceptor cells of the retina (Figure 6C), whereas gustducin anti-sense probe did not (Figure 6D). Nevertheless, because gustducin target mRNA is highly abundant in taste bud cells and because RNAs for the two proteins share sequence identity, it was important to ascertain the extent to which the transducin probe might crossreact with gustducin mRNA. We measured this crossreactivity by running a Northern blot loaded with equal amounts of in vitro-transcribed sense RNAs for transducin and gustducin. The blot was hybridized with DIG-labeled probes at high stringency (52C), very similar to the stringency in ISH. As expected, transducin probe hybridized strongly to its complementary target (transducin sense RNA; Figure 6E). The autoradiographic signal for hybridization to an equal quantity of gustducin target was less than 1% that for transducin target RNA.
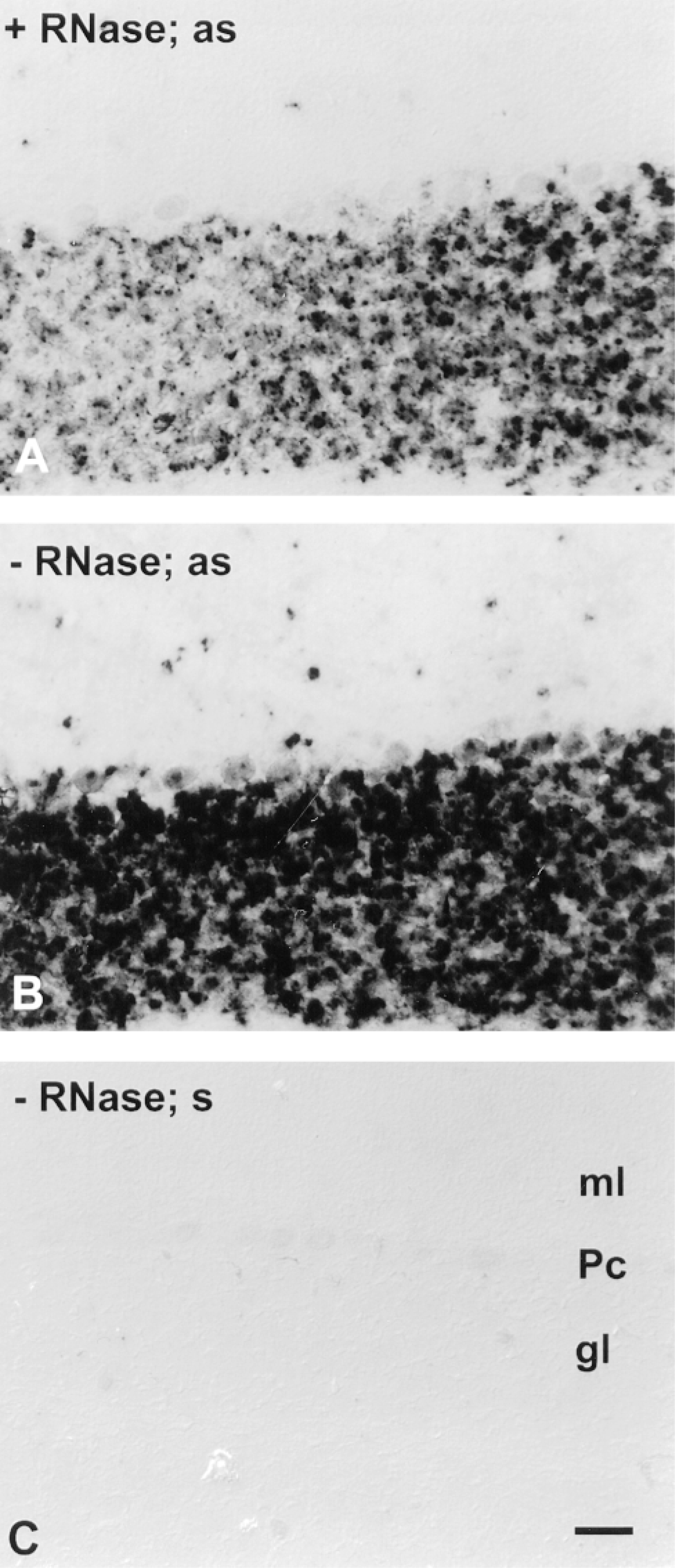
RNase digestion after hybridization is unnecessary and may be deleterious. Cryosections of cerebellum were pretreated (including proteinase K) and were hybridized at high stringency with 50 ng/ml of 2.6-KB probes for mGluR4. After hybridization, sections were treated with RNase (A) or were not treated with RNase (B,C). The color development reaction proceeded for 1 hr for each section. Hybridization signal was detected by the TSA-AP protocol and is visible in sections hybridized with anti-sense (as) probe (A,B) in the granule cell layer (gl) but not appreciably in the molecular layer (ml). The level of nonspecific staining, e.g., as seen in Purkinje cells (Pc), is similar whether RNase digestion was employed or not and is similar to the level of nonspecific staining in sections hybridized with sense (s) probe (C). Bar = 40 μm.
The ISH results showed a clear difference in the numbers of taste bud cells stained for gustducin and transducin. To obtain a preliminary estimate of the relative incidence of taste bud cells expressing these G-proteins, we counted labeled cells from sections hybridized with the two probes in parallel. For this analysis, we used identical hybridization and detection parameters that gave the strongest signals to ensure detection of all transducin-positive cells. Only taste buds in which a taste pore was clearly visible were included in these measurements. We analyzed about 150 taste buds for each probe (two separate experiments). On average, 1.2 cells were counted as transducin-positive and 6.2 cells were gustducin-positive per taste bud profile in 10-μm cryosections of adult rat vallate and foliate papillae, i.e., approximately five times as many gustducin-positive as transducin-positive cells were visible in taste buds.
Hybridization of transducin probe to gustducin target is at least 100 times less effective than to transducin target mRNA (Figure 6E). Therefore, we conclude that our hybridization signals with TSA-AP most likely represent genuine transducin mRNA in taste bud cells.
mGluR4 mRNA Is Found in Some Taste Bud Cells. Chaudhari et al. (1996) showed by in situ hybridization with 33P-labeled probes that a metabotropic glutamate receptor, mGluR4, is expressed at low concentration in taste buds but not in surrounding nonsensory epithelium. The resolution afforded by radiolabeled probes did not permit cellular localization of the hybridization signal and therefore did not enable us to estimate the number of taste bud cells that expressed this mRNA. Therefore, we applied TSA-AP for mGluR4 in lingual epithelium to improve the ISH resolution and extend our earlier findings.
Figure 7A shows hybridization with mGluR4 anti-sense probe (2.61 KB) in taste bud cells from a 24-day-old rat vallate papilla sectioned on a vibratome. The signal appears as crescent-shaped depositions of chromogen in the perikaryon, with occasional punctate staining in the elongate apical and basal processes of individual taste bud cells. Control hybridization with sense probe in adjacent sections gave no staining (Figure 7B). Similar results were obtained with 10-μm cryosections. However, with 40-μm vibratome sections signal could be visualized throughout entire cells. We have observed similar results in 50 sections in at least six independent experiments in foliate and vallate papillae.
As shown in Figure 7A, only a few taste bud cells displayed signal. A rough estimate of the incidence of mGluR4 expression, obtained from counting stained cells in 40-μm vibratome sections, gave an average of 1.8 cells per vallate taste bud profile in 18–24-day-old rats (total 134 buds counted). Even with TSA-AP, we may be at the limit of detection for mGluR4, and therefore the incidence of cells expressing this receptor may well be underestimated. Sections hybridized with gustducin probe in parallel and under identical conditions contained 4.2 stained cells per taste bud profile (total 38 buds counted).
Discussion
The success of ISH for rare mRNAs depends strongly on both the integrity of target mRNAs in sections and the ability of probes to penetrate the sections and hybridize with mRNAs. Another requirement is a powerful reporter system capable of revealing low numbers of probe-mRNA hybrids per cell while keeping background staining as low as possible. Several methods have been published for signal amplification in immunohistochemistry and ISH. Examples include sandwich protocols with avidin-biotin-peroxidase or peroxidase-anti-peroxidase complexes, as well as silver intensification of chromogen deposits (Liposits et al. 1991; Gallyas et al. 1993). However, in many cases, amplification protocols also increase nonspecific background staining, thereby limiting their utility for visualizing rare mRNAs. The protocol described here optimizes signal-to-noise ratio by improving mRNA preservation, optimizing hybridization conditions, and introducing TSA-AP, a potent signal amplification system.
Fixation and Permeabilization Must Be Adjusted for Each Tissue Type
To detect rare messages, it is especially important to preserve the integrity of target mRNAs. With lingual tissue, we found that cryosections could not be stored at −80C for more than 2 days before loss of signal intensity became noticeable. In addition, we found that rapid fixation at 4C enhanced signal intensity for rare mRNAs. We interpret these results to reflect a high endogenous content of RNase in lingual tissue that easily compromises target mRNA integrity. Similar precautions may be necessary for other tissues as well.
It is also critical to permeabilize the sections adequately. We found that brain and lingual tissues were best permeabilized with a brief proteinase K digestion, whereas detergent treatment was much less effective. The benefit of proteinase treatment depends on tissue type and on fixation conditions (Singer et al. 1986; Larsson and Hougaard 1990; Wilkinson and Nieto 1993). Permeabilization with detergents is necessary when tissues rich in lipids are used, e.g., myelinated brain regions (Singer et al. 1986; Schaeren-Wiemers and Gerfin-Moser 1993; Panoskaltsis-Mortari and Bucy 1995). When ultrastructural morphology is to be preserved or when cell cultures are used, a mild detergent pretreatment appears to be more effective than proteinase digestion (Yamawaki et al. 1993; Olink-Coux and Hollenbeck 1996; Prakash et al. 1997). In some cases, sufficient ISH signal is obtained when the probe is applied to tissues without any permeabilization at all (Wahle and Beckh 1992; Litman et al. 1993,1994). These examples show that permeabilizing the samples must be adjusted for each tissue type.
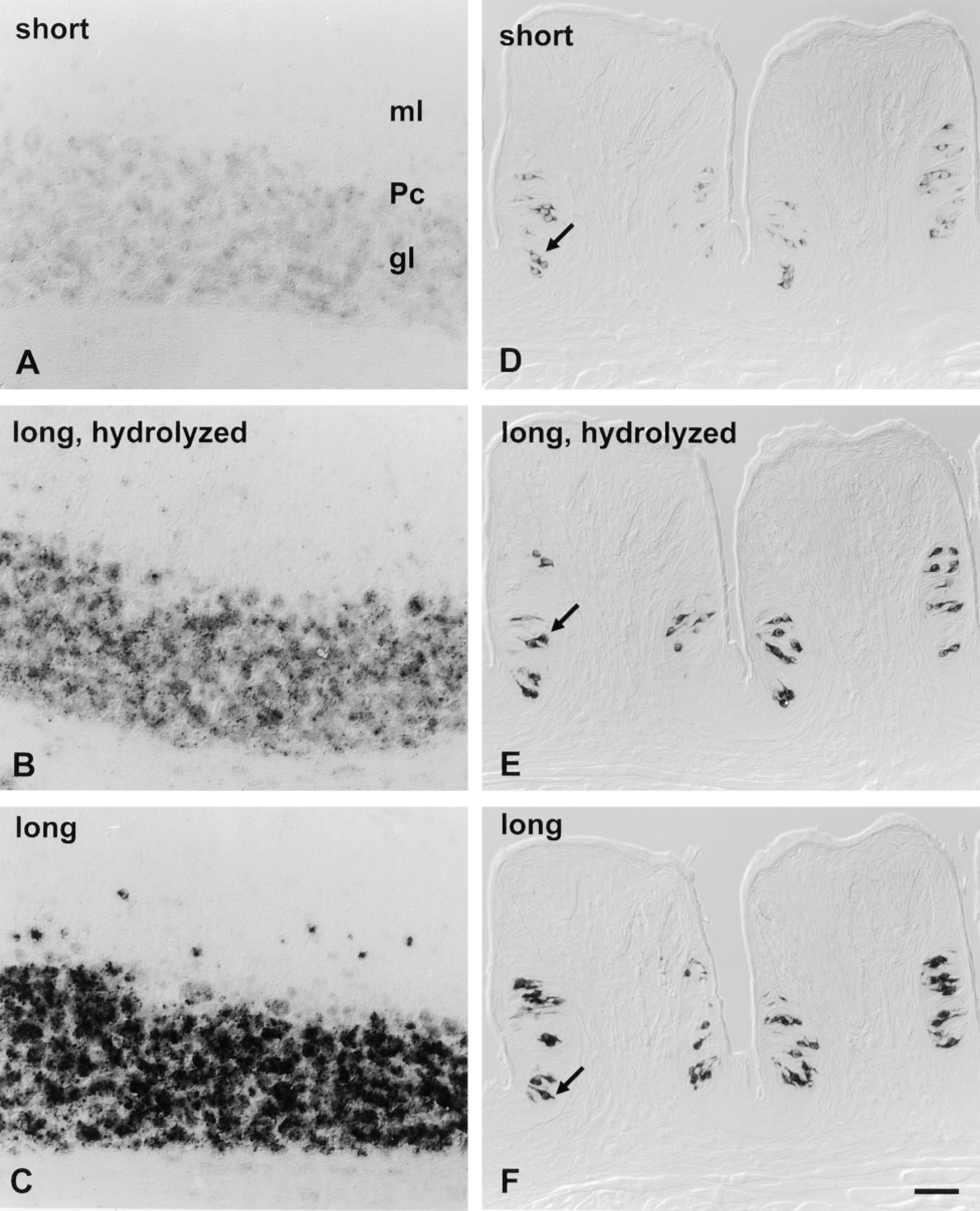
Hybridization signal intensity is increased with more complex and longer probes. Cryosections of cerebellum were hybridized in parallel with 0.8-KB i.e., “short” anti-sense probe (A), 2.6-KB “long” anti-sense probe hydrolyzed to a mean length of 0.3-KB (B), or intact 2.6-KB, i.e., “long” anti-sense probe for mGluR4 (C). Cryosections of foliate papilla were hybridized in parallel with 0.27-KB, i.e., “short” anti-sense probe (D), 1.37-KB, i.e., “long” anti-sense probe hydrolyzed to a mean length of 0.3-KB (E), or intact 1.37-KB “long” anti-sense probe for gustducin (F). Experimental conditions included proteinase K predigestion and no RNase digestion after hybridization. Color development reaction times for cerebellar sections with TSA-AP were 1 hr (A-C), whereas foliate hybridizations were visualized with the unamplified AP protocol for 2.5 hr (D-F). For each series, signal intensity increases with probe complexity (A vs B and D vs E) and with physical length of probe (B vs C and E vs F). In each case, signal is visible only in the appropriate cells (as described in Figures 2 and 4). Bar = 40 μm.
Longer ISH Probes Generate Stronger Signals
Conventional wisdom suggests that decreasing the length of ISH probes improves their penetration into tissue sections. Partial alkaline hydrolysis has been recommended to reduce long probes into shorter fragments 0.2–0.4 KB in length (Angerer and Angerer 1992; Schaeren-Wiemers and Gerfin-Moser 1993). On the contrary, we found that a stronger signal was achieved using complex probes that were unfragmented. A mixture of short probes generated by partial hydrolysis of complex probes (and which thereby covered an equivalent region of target mRNA) yielded a weaker signal. These results suggest that long probes can form more stable hybrids than a mixture of short probes. The enhancement of signal through increased probe complexity is intuitive and well known, although the negative impact of fragmentation has not been documented.
Posthybridization Treatment with RNase Can Decrease ISH Signal
An RNase treatment is recommended in several ISH protocols to decrease nonspecific background (Wahle and Beckh 1992; Litman et al. 1993; Panoskaltsis-Mortari and Bucy 1995; Prakash et al. 1997). However, we found that RNase treatment after hybridization was not necessary and, in fact, markedly reduced the signal. In our experience, nonspecific background is best controlled by maintaining high stringency during hybridization and washing rather than through RNase digestion.
Tyramide Signal Amplification Is Improved by Decreasing Probe Concentration
A chromogenic ISH protocol with strong signal amplification is needed to detect rare mRNAs in cells. Tyramide signal amplification (TSA) is an effective intensification procedure in peroxidase-based immunodetection assays. The procedure employs peroxidase to catalyze the deposition of biotinylated tyramides (Bobrow et al. 1992). TSA has been reported to give a remarkable increase in sensitivity in immunohistochemistry and immunoblots (Hunyady et al. 1996; Schöfer et al. 1997). TSA has also been used for fluorescent in situ hybridization (FISH) on chromosomes (Kerstens et al. 1995; Raap et al. 1995; de Haas et al. 1996; Macechko et al. 1997; Speel et al. 1997; van Gijlswijk et al. 1997). TSA has only recently been modified to visualize mRNA in tissue sections and cells (Wanner et al. 1997; Schmidt et al. 1997; Landry and Hökfelt 1998).
A limitation on the use of TSA for rare mRNAs is the problematic rise of nonspecific background staining. However, our results show that a crucial improvement in signal-to-noise ratio can be achieved by lowering probe concentration about tenfold from concentrations recommended in many protocols (e.g., Schaeren-Wiemers and Gerfin-Moser 1993; Guiot and Rahier 1995). A probe concentration of 10–30 ng/ml is saturating, as has been previously estimated (Singer et al. 1986), and does not limit specific signal. This implies that signal intensity is determined by the abundance of the target mRNA rather than by probe concentration. In contrast, nonspecific background appears to be determined by probe concentration.
Summary of steps for ISH and detection using the TSA-AP protocol

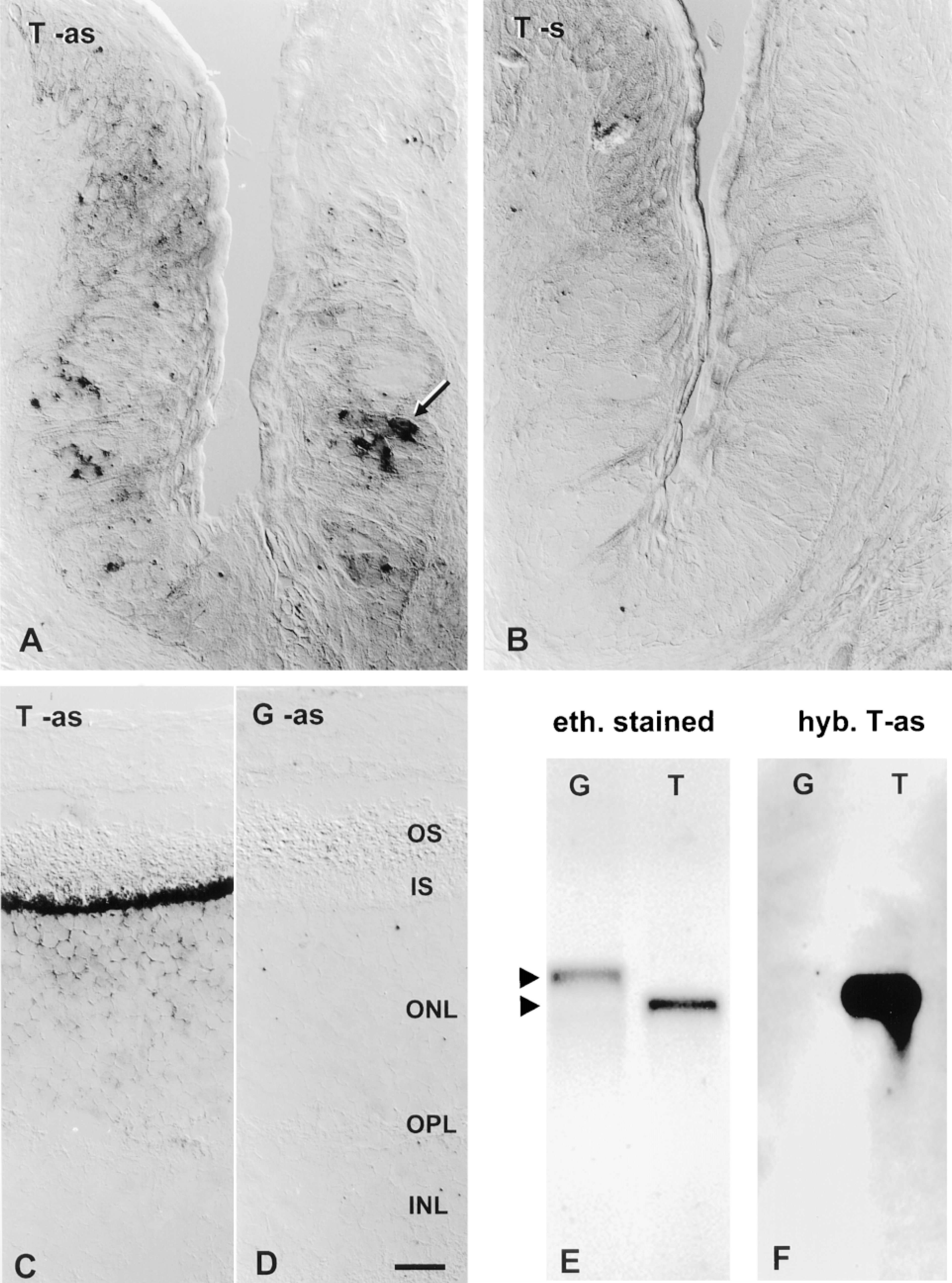
ISH with transducin probe in taste buds and retina. Cryosections from foliate papilla were hybridized in parallel with 1.03-KB DIG probes for transducin. Hybridization signal is seen in some taste bud cells (arrows) hybridized at high stringency with anti-sense (as) probe (A) but not in sections hybridized with sense (s) probe (B). As expected, transducin anti-sense probe gave a strong signal in the photoreceptor cells of the retina (C), whereas gustducin 1.37-KB anti-sense probe did not (D). Transducin signal in the retina is limited to the inner segment (IS) and outer nuclear layer (ON) of photoreceptors and is absent from the outer segment (OS), outer plexiform layer (OPL), and inner nuclear layer (INL). Experimental conditions for ISH included treatment with proteinase K and no RNase digestion. Color development reaction times were up to 60 min for TSA-AP (A,B) and 2 hr for unamplified protocol (C,D). Bar = 20 μm. (E) One hundred ng each of sense RNA for gustducin (G) or transducin (T) was electrophoresed and visualized by ethidium bromide before transferring to nylon membrane. (F) The resulting Northern blot was hybridized at equivalently high stringency (as for ISH above) with anti-sense probe for transducin (T-as). A faint cross-hybridization signal was seen for the gustducin target band and was quantified by densitometry.
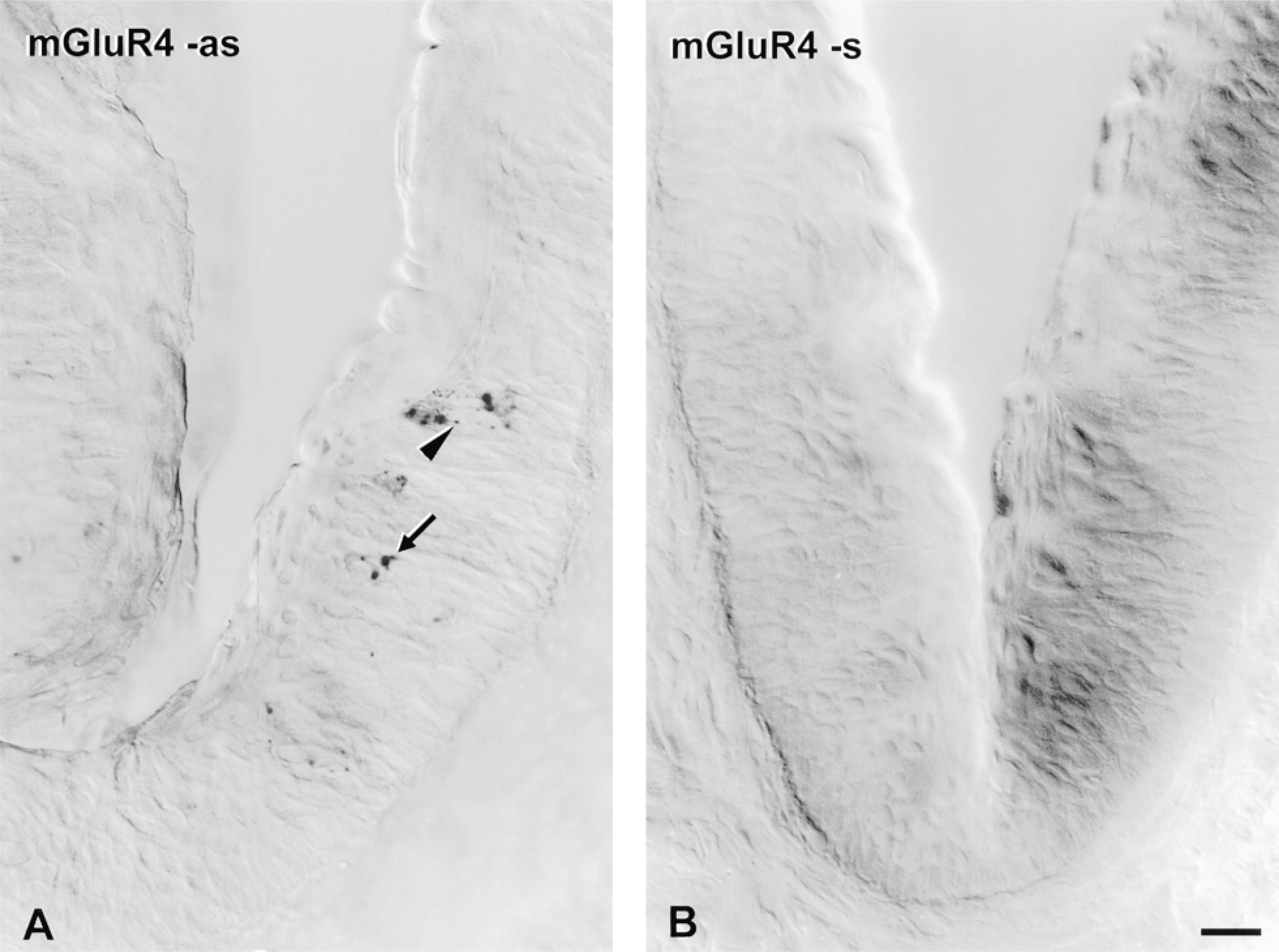
ISH with mGluR4 probe in taste buds. Vibratome sections of vallate papilla were hybridized in parallel with 2.6-KB DIG probes for mGluR4. The color development reaction was allowed to proceed for 1 hr for each section. Hybridization signal is visible in the perikarya (arrow) of some taste bud cells and also appears as punctate deposits in elongate apical or basal processes (arrowhead) of taste bud cells hybridized with anti-sense (as) probe (A). No dark deposits were visible in surrounding nonsensory epithelium or in underlying tissues. No signal was visible in taste bud cells or other cell types after hybridization with sense (s) probe (B). Bar = 20 μm.
Signal Transduction in Taste Receptor Cells: Receptors and G-proteins
Recent advances in molecular biology have begun to identify the proteins that generate the initial events in chemosensory transduction in taste buds. In the case of taste stimuli that are transduced by receptors coupled to intracellular second messenger cascades, no receptors uniquely expressed in taste bud cells have been cloned. Nevertheless, we have previously demonstrated that mGluR4 is expressed in taste buds and may function as a taste receptor for the transduction of glutamate (Chaudhari et al. 1996; Bigiani et al. 1997). Our earlier studies indicated that mGluR4 mRNA was present in very low abundance in taste buds. However, with 33P-labeled probes it was not possible to determine whether all or only a subset of taste bud cells expressed this receptor. With the TSA-AP detection described in this report, we observed an unambiguous and specific hybridization signal for mGluR4 mRNA in limited numbers of single cells in taste buds from circumvallate and foliate papillae (Figure 7). The following findings indicate the specificity of the mGluR4 ISH signal: (a) sense probe did not label cells in sections adjacent to those hybridized with anti-sense probe; (b) anti-sense probe yielded staining only in the perikarya of taste bud cells organized within taste buds and not in other cell types in the adjacent nonsensory epithelium or in connective tissue. Anti-sense probe produced punctate staining also in the apical and basal processes of elongate taste bud cells. Only one or two cells per taste bud were labeled with mGluR4 anti-sense probe, and not all taste buds contained labeled cells. The low incidence of taste bud cells expressing mGluR4 is consistent with estimates of the low abundance of mRNA for this receptor in taste buds, based on RNase protection assays with RNA extracted from tissue (Chaudhari et al. 1996).
Transducin mRNA has been estimated, using RNase protection assay, to be about 25 times less abundant than gustducin mRNA in taste buds (Ruiz-Avila et al. 1995). Our results are consistent with these findings, i.e., the transducin signal was considerably weaker than signal with gustducin probe in taste buds and required longer color development to visualize the signal. Furthermore, ISH with transducin probe yielded a signal only in a small fraction of taste receptor cells. We estimated that there are four to seven times fewer cells expressing transducin than gustducin. The distinctly different number of intensely stained taste bud cells for the two probes suggests that transducin probe did not crossreact with gustducin mRNA target under the stringent hybridization conditions used in our study. A detailed morphometric analysis of gustducin immunocytochemistry has indicated that there are about nine or ten gustducin expressing cells per taste bud in rat circumvallate and foliate papillae (Boughter et al. 1997). On the basis of that figure and our own estimates, we suggest that on average there are between one and three cells per taste bud that express transducin. Further investigations using double-labeling ISH will be necessary to show whether gustducin and transducin are co-expressed in distinct taste bud cells.
A number of putative components of sensory transduction pathways in taste buds have been identified by cDNA cloning (McLaughlin et al. 1992,1994; Li et al. 1994; Chaudhari et al. 1996; Misaka et al. 1997). Functional assays performed in vitro by reconstituting membrane fractions and G-proteins have implicated transducin and gustducin in sensory transduction for sweet and bitter taste stimuli (Ruiz-Avila et al. 1995; Wong et al. 1996; Ming et al. 1998). Nevertheless, to establish unequivocally the physiologically relevant combinations of receptors, G-proteins, and effectors in situ, it will be necessary to demonstrate their coexistence in individual taste bud cells. Double-label immunocytochemistry or ISH could address this. However, demonstrating a lack of crossreactivity of antibodies against closely related proteins is more laborious than controlling crossreactivity of RNA probes by stringency in ISH. Therefore, ISH with a highly sensitive detection system, such as the TSA-AP that we have described here, can potentially play a vital role in elucidating taste transduction mechanisms.
Footnotes
Acknowledgements
Supported by a grant from NIH, NIDCD DC03013.