
Abstract
Keywords
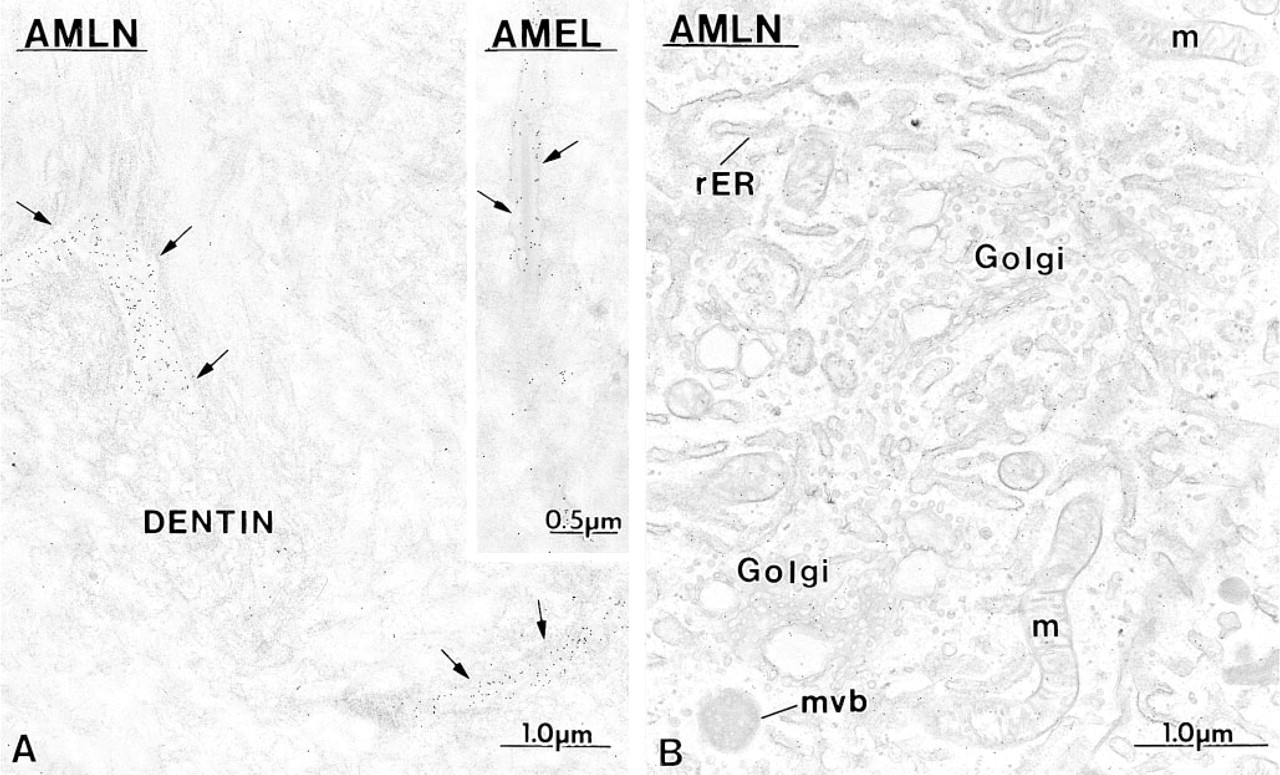
A
During appositional growth of the enamel layer, secretory granules in ameloblasts are characteristically routed towards two spatially distinct secretory sites, at which they release their contents constitutively to build up interrod and rod areas and, hence, bulk enamel thickness (Nanci and Warshawsky 1984). The organic phase of developing enamel in optimally fixed teeth appears morphologically homogeneous by electron microscopy. This suggests that constituent amelogenins and various anionic proteins may be uniformly spread, as intact proteins or fragments, throughout the thickness of the enamel layer, albeit in decreasing total bulk amounts forward in time as enamel matures (discussed in Nanci et al. 1996). There are several lines of evidence suggesting that this is not the case. Both newly secreted matrix proteins present at the forming surface of the enamel layer and partially degraded molecular forms located deeper into it can be concentrated at, or relatively missing from, specific sites within the layer (Smith et al. 1989a,b; Kogaya 1994; Nanci et al. 1996; Murakami et al. 1997; Uchida et al. 1991a,b,1997). The most dramatic differences in intraenamel protein concentrations have been observed for nonamelogenins such as ameloblastin and enamelin, for which newly secreted forms are present in high concentration at the enamel surface near ameloblasts (Hu et al. 1997a,b; Murakami et al. 1997; Uchida et al. 1997).
The spatial relationship between amelogenins and ameloblastin, both when these proteins are secreted and after postsecretory processing, has not been examined in any detail. The purpose of this investigation was to obtain information about the intracellular and extracellular distribution of ameloblastin vs amelogenin using qualitative and quantitative high-resolution immunocytochemistry in combination with immunoblotting and polyclonal antibodies against the full-length and an internal portion of recombinant ameloblastin, in contrast to synthetic peptide antibodies as was employed in a recent study reported by Uchida et al. (1997). Another objective was to clarify issues about sites of expression vs sites of secretion for ameloblastin. It is presently unclear at what point in development ameloblastin first appears extracellularly and at what point this nonamelogenin is no longer secreted from ameloblasts.
Materials and Methods
Antibodies and mRNA Probes
Polyclonal rabbit antibodies against (a) recombinant M179 mouse amelogenin (AMEL, equivalent to the main M180 isoform minus the N-terminal methionine group and lacking a phosphate group on SER16; Simmer et al. 1995), (b) recombinant rat amelin 1 (AMLN, full-length; Fong et al. 1996b), and (c) recombinant rat ameloblastin (AMBN, internal portion of the molecule extending from residues 175 to 348; Krebsbach et al. 1996) were prepared and purified as previously described. For mRNA probes, inserts of ameloblastin DNA (1900
Tissue Preparation
All animal handling and experimental procedures were approved by the Comité de Déontologie de l'Expérimentation sur les Animaux of the Université de Montréal. Male Wistar rats weighing ~100 g were used for all analyses (Charles River Canada; St-Constant, QC, Canada).
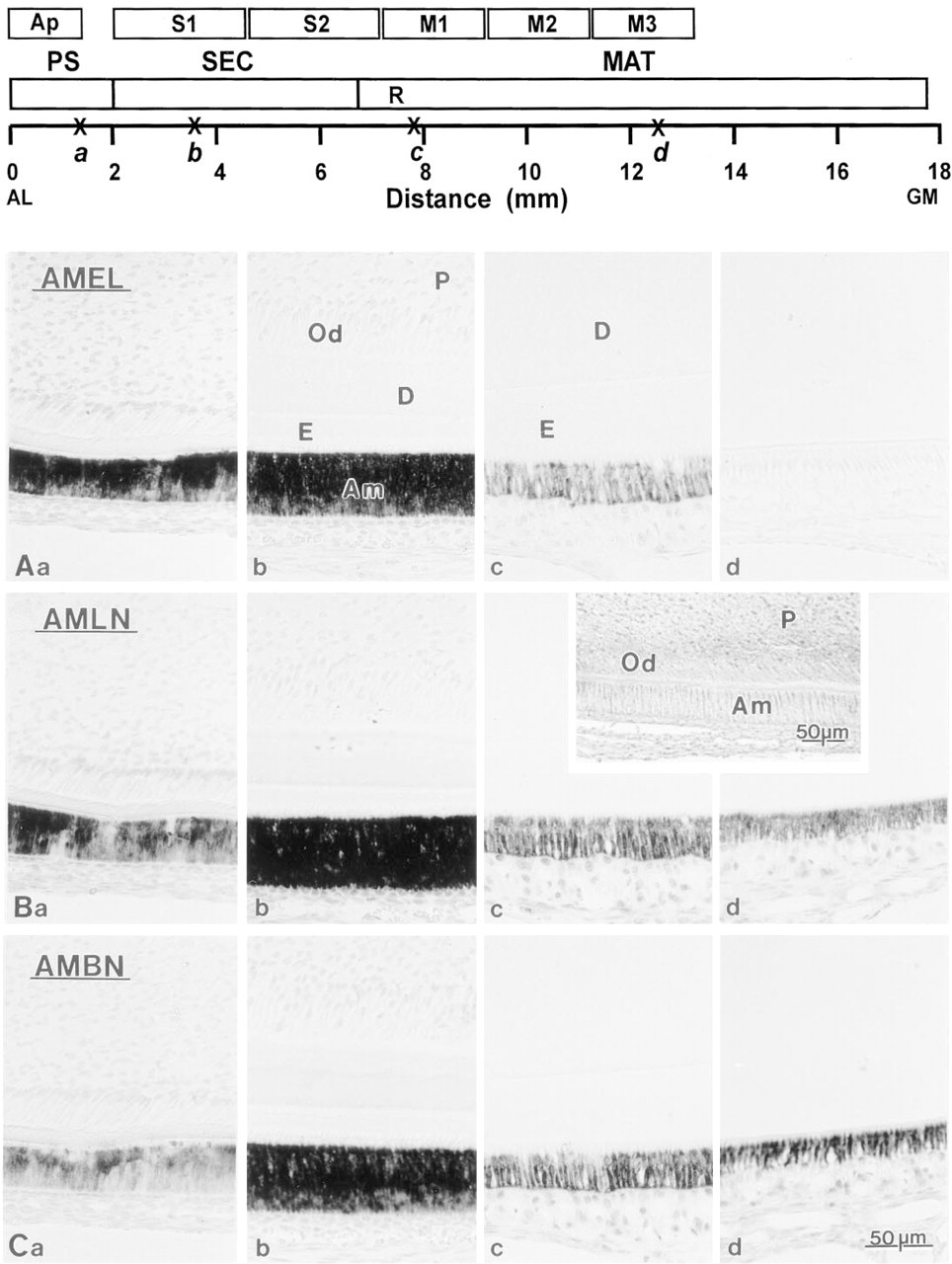
Comparative photomicrographs illustrating in situ hybridization reactions with probes for recombinant amelogenin (AMEL;
Biochemical Analyses
Some rats were anesthetized with diethyl ether and immediately decapitated. Others were anesthetized with chloral hydrate (0.4 mg/g bw), injected
Histological Studies. Rats were anesthetized with chloral hydrate (0.4 mg/g bw) and perfused for 30 sec by a cannula inserted through the left ventricle into the ascending aorta with lactated Ringer's solution (Abbott), followed for 20 min by either 4% paraformaldehyde in 0.01 M PBS, pH 7.2 (for in situ hybridization studies) or 4% paraformaldehyde +0.1% glutaraldehyde or 1% glutaraldehyde in 0.08 M sodium cacodylate containing 0.05% CaCl2, pH 7.2 (for immunocytochemical studies). Some rats were treated with cycloheximide for 6 hr or infused with brefeldin A (Sigma) for 1 hr as previously described (Nanci et al. 1996; Hashimoto and Nanci 1996) before being perfused. After perfusion, the hemimandibles were removed from each rat and further fixed by immersion in the same solution at 4C for an additional 3-18 hr. The hemimandibles were then washed in their respective fixative buffer and decalcified for 14 days at 4C in 4.13% EDTA (Warshawsky and Moore 1967). For in situ hybridization they were dehydrated in graded ethanols and processed for embedding in paraffin. The remaining decalcified hemimandibles were cut into segments from the presecretory, early secretory, and early to midmaturation stages using a molar reference line (illustrated in Figure 1; Smith and Nanci 1989a). Some segments were postfixed with potassium ferrocyanide-reduced osmium tetroxide (Neiss 1984), and others were left unosmicated. All specimens were dehydrated in graded alcohols and processed for embedding in LR White resin (London Resin; Berkshire, UK) as described previously (Bendayan et al. 1987; Nanci et al. 1989). Thin sections (~100 nm) were cut with a diamond knife, mounted on Formvar-carbon-coated nickel grids, and processed for postembedding protein A-gold immunocytochemistry (reviewed in Bendayan 1995). In all cases, grids with sequential sections from a block were incubated with the various antibodies, allowing a relatively direct comparison of their labeling patterns.
Immunoblotting
Thirty μl of extraction fluids from each vial was applied to separate lanes of standard format (16 cm × 14 cm × 1 mm) 12% polyacrylamide slab gels along with at least one lane of broad range molecular weight marker proteins (Bio-Rad; Mississauga, ON, Canada) and 1 μg per lane of purified rat serum albumin (Sigma). Proteins were separated by electrophoresis at 20 mA per gel constant current using a discontinuous buffer system (Laemmli 1970). They were then electrotransferred from the gels onto 0.45-μm pore size nitrocellulose membranes and probed with primary and alkaline phosphatase-labeled secondary antibodies as described previously (Chen et al. 1995).
In Situ Hybridization
Sections of the entire hemimandible were cut at 5-10-μm thickness and mounted on aminoalkylsilane-coated glass slides (Sigma). The paraffin was removed with xylene and the sections were rinsed in PBS and treated at 37C with proteinase K (20 μg/ml; Boehringer Mannheim) for 30 min in a buffer consisting of 100 mM Tris-HCl and 50 mM EDTA, pH 8.0. After digestion they were rinsed in 0.2% glycine, fixed with 4% paraformaldehyde in PBS for 5 min, and immersed for 10 min in 20 mM triethanolamine containing 0.5 ml concentrated acetic anhydride. The slides were then rinsed with PBS and treated with a prehybridization solution consisting of SSC (300 mM NaCl + 30 mM sodium citrate) 2 × containing 50% deionized formamide, for 60 min at 50C. Hybridization was carried out by incubating the slides overnight at 50C in a humidified chamber with 50% formamide, 2 × SSC, 1 × Denhardt's solution, 10% dextran sulfate, 500 μg/ml herring sperm DNA, and 250 μg/ml yeast tRNA containing ~0.5 ng/μl of the anti-sense or sense probe. After hybridization they were washed several times with 4 × SSC. Nonhybridized transcripts were digested for 30 min at 37C with 20 μg/ml RNase A (Boehringer Mannheim) in 500 mM NaCl, 10 mM Tris-HCl, 1 mM EDTA, pH 8.0. Digested sections were washed with decreasing concentrations of SSC (× 4, 2, 1, and 0.1) for 30 min each at 4C. The hybridized probe was then detected by incubating with a sheep anti-digoxigenin antibody conjugated to alkaline phosphatase (Boehringer Mannheim) for 2 hr at 4C. Phosphatase activity was revealed with 450 μg/ml nitroblue tetrazolium and 175 μg/ml of 5-bromo-4-chloro-3-indolyl phosphate in 100 mM Tris-HCl, pH 9.5, containing 100 mM NaCl and 50 mM MgCl2.
Postembedding Colloidal Gold Immunocytochemistry
Sections of osmicated samples were first treated with 5% sodium metaperiodate for 60 min (Bendayan and Zollinger 1983), and unosmicated ones were directly processed for immunolabeling. The sections were floated for 5 min on a drop of 0.01 M PBS containing 1% ovalbumin (Oval), pH 7.4, and transferred onto a drop of rabbit anti-amelin (diluted 1:200), anti-ameloblastin (diluted 1:20), or anti-amelogenin (diluted 1:300) antibody for 1 hr. After incubation the grids were washed by floating on PBS, again placed on a drop of PBS-Oval for 5 min and transferred onto a drop of protein A-gold complex. The complex was prepared as described in Bendayan (1995) using colloidal gold particles of 10-12 nm (Frens 1973). The grids were jet-washed with PBS followed by dH2O, stained with uranyl acetate and lead citrate, and examined by transmission electron microscopy using a JEOL JEM 2000FX-II operated at 80 kV. Controls for the specificity of the labeling consisted of incubating the sections with preimmune sera, rabbit antibodies to unrelated proteins, or with protein A-gold alone.
Quantification of Immunolabeling
Incisors from three rats perfused with 4% paraformaldehyde +0.1% glutaraldehyde were used for quantitative analyses. Consecutive thin sections were incubated with one of the three antibodies and random fields showing the apical membrane of ameloblasts and adjacent enamel layer were photographed in the electron microscope from which prints at a final magnification of × 30,000 were made. A rectangle representing 250 nm height × 1000 nm width (250,000 nm2) was superimposed over the region of enamel immediately adjacent to the apical plasma membrane of ameloblasts (Window 1), and the number of gold particles falling within the rectangle was scored (see Figures 9 and 15). The rectangle was then moved in steps away from this first position to create a series of 12 consecutive counting windows. This procedure was followed to quantify areas of (a) initial enamel (secretory stage), (b) forming inner enamel (interrod and rod; secretory stage), and (c) final enamel (very early maturation stage). Areas of forming interrod enamel extending as prongs along the sides of forming enamel rods (secretory stage) were narrow and had to be quantified with a window having one half the area of the larger window (250 nm height × 500 nm width = 125,000 nm2). Data were entered into Version 5.1G of STATISTICA for Windows (Statsoft; Tulsa, OK) coded by photograph, antibody, window location, number of gold particles for the window, and total counts per strip of windows in a group. Final data were expressed on a relative basis as the percent total number of gold particles per strip of 12 windows for each antibody. An average of 355 total windows was counted per antibody, yielding a minimum of approximately 30 samples per window location for a given group of 12 windows. Significance tests on raw particle counts were done by two-way analysis of variance using antibody type (three total) and window location (12 total) as grouping variables for each sampling region analyzed.
Results
Global Distribution of mRNA Signals and Translated Enamel Matrix Proteins
In Situ Hybridization. Incubation of incisor sections with anti-sense mRNA for amelogenin and ameloblastin/amelin resulted in staining that extended from the presecretory to the maturation stages of amelogenesis (Figure 1). With all three probes used, mRNA expression was seen to start before dentin formation. The signals increased gradually, were most intense during the secretory stage, and diminished in the maturation stage. Expression of amelogenin mRNA was not discernible by midmaturation (Figure 1a-d), whereas the ameloblastin/amelin mRNA signals were still intense at midmaturation (Figures 1Bd and 1Cd) and persisted at moderate to low levels into the late maturation stage. Control incubations with sense probes showed no staining over ameloblasts (Figure 1 inset).
Immunoblotting. Antibodies to full-length (anti-amelin) and the middle portion (anti-ameloblastin) of ameloblastin reacted with several groups of proteins in whole enamel organ cell and enamel homogenates that were distinctly different in molecular weight compared to proteins immunostained by anti-amelogenin at all locations on the tooth (Figures 2 and 3). As shown previously (Simmer et al. 1994), the anti-amelogenin antibody reacts with various protein bands, including the native protein, its isoforms, and their degradation products (Figure 2). In some cases similar proteins were revealed by both anti-amelin and antiameloblastin antibodies, whereas others appeared immunoreactive to only one of the antibodies (Figures 2 and 3). Whole apical segments showed fairly strong immunostaining of proteins near 27 kD with anti-amelogenin and near ~58 kD with anti-amelin and anti-ameloblastin (Figure 2). Secretory (S1 and S2) and early maturation (M1) stage cell homogenates typically showed two clusters of highly anti-amelin-reactive proteins near 66 and 58 kD and several more weakly immunostained proteins between 50 and 21 kD (Figure 3B). These proteins were still evident during the early maturation stage, but the overall intensity of immunostaining weakened in an incisal direction (M1 > M2 > M3; Figure 3B). Results were similar for anti-ameloblastin, except that cell proteins below 58 kD were generally weakly immunostained (Figure 3C). Immunostaining of proteins near 58 kD was weak in maturation stage samples with anti-ameloblastin and was generally restricted to M1 samples (Figure 3C). Enamel homogenates from secretory and early maturation stage showed two highly immunoreactive proteins near 65 and 50 kD with both anti-amelin and anti-ameloblastin antibodies (Figures 3B and 3C). In addition, anti-amelin consistently immunostained several proteins near 50-31 kD and it strongly immunostained a group of proteins between 21 and 14 kD, some of which were not usually revealed by antiameloblastin (Figures 3B and 3C). Weak staining of these lower molecular weight proteins was still seen in midmaturation stage samples with anti-amelin (Figure 3B). Rats treated for 2 hr with cycloheximide showed marked changes in banding patterns for some intra-cellular and extracellular proteins most noticeably near 65 kD with anti-amelin and near 65, 58 (in cells), and 50 (in enamel) kD with anti-ameloblastin (Figures 3A-3C). Changes in banding pattern persisted in enamel at 6 hr after cycloheximide administration but 65- and 58-kD bands in cells began to reappear at this interval (data not shown).
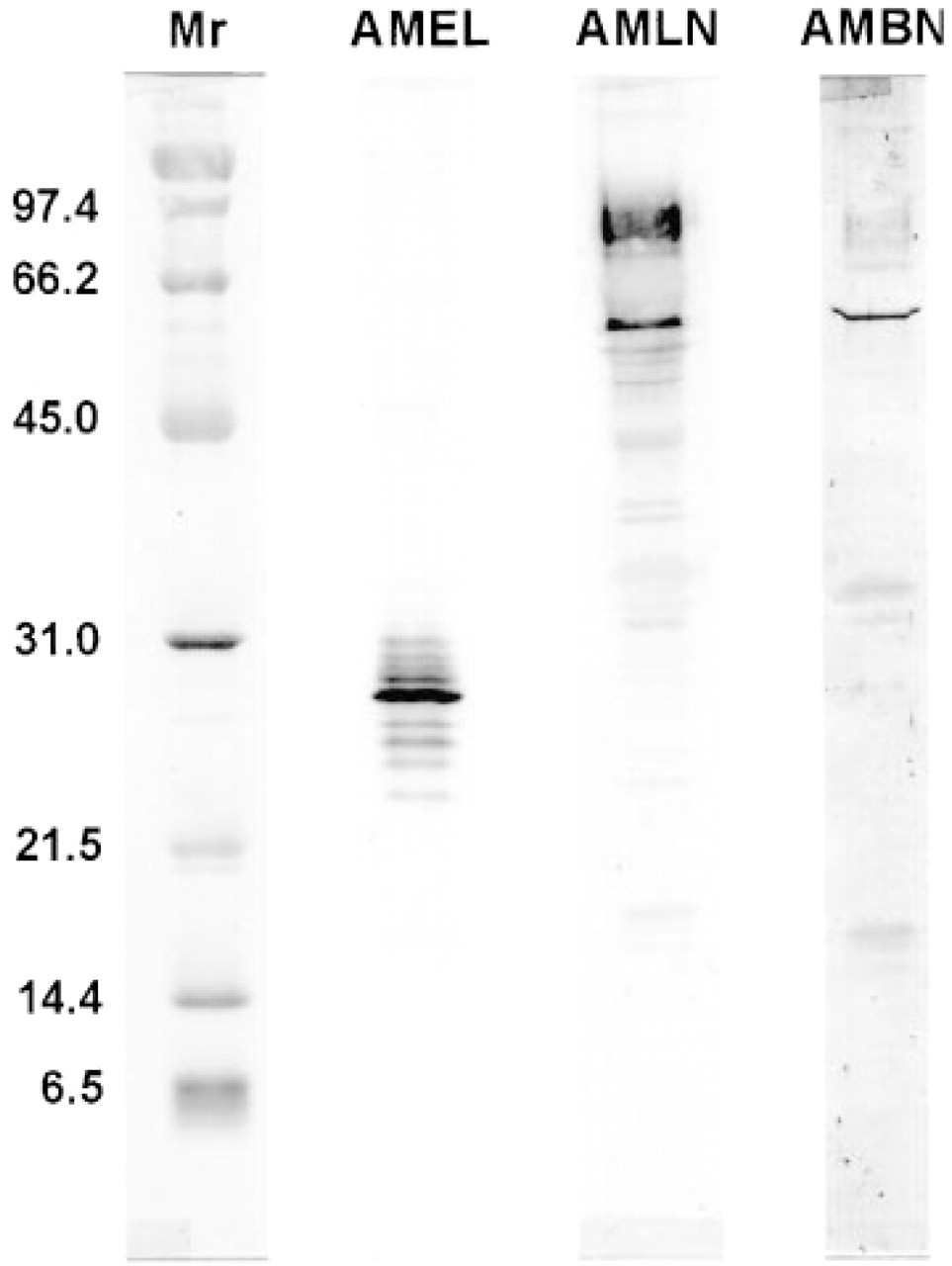
Immunoblots of apical segments from rat incisors (see drawing in Figure 1 for orientation) probed with anti-amelogenin (AMEL), anti-amelin (AMLN), and anti-ameloblastin (AMBN) antibodies. The column at the left shows standard broad-range molecular weight marker proteins (Mr) stained with Ponceau S (Coudrier et al. 1983). Presecretory stage samples show staining for both amelogenin and ameloblastin. Note, however, the absence of some lower molecular weight bands seen in older enamel samples stained with both antibodies to ameloblastin (see Figure 3).
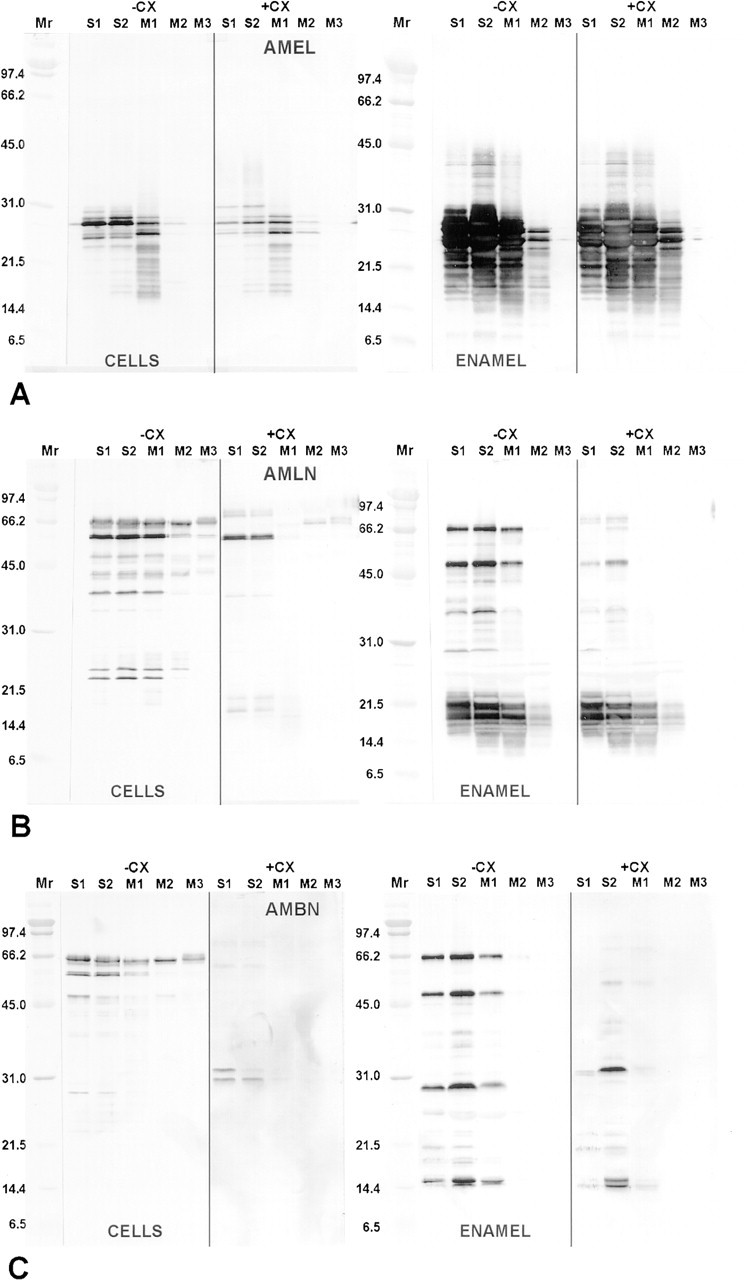
Immunoblots of whole enamel organ cell (CELLS) and enamel homogenates (ENAMEL) from normal rats (-CX) and those treated with cycloheximide (+CX) for 2 hr probed with anti-amelogenin (AMEL)
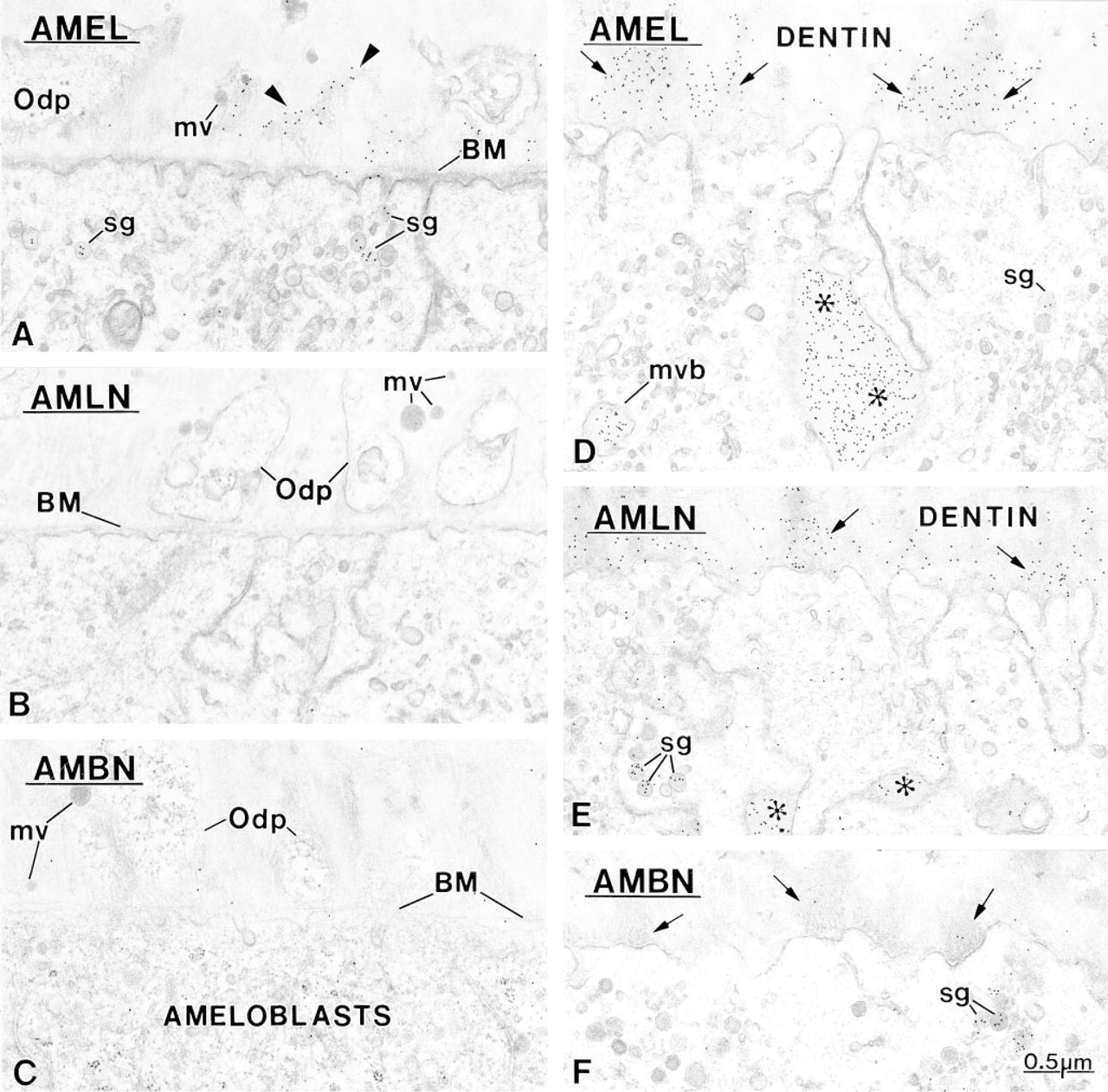
Amelogenin (AMEL) is immunodetected in the forming extracellular matrix before ameloblastin. Initially, amelogenin (arrow-heads) is associated with the lamina fibroreticularis of the basement membrane (BM) separating the differentiation odontoblasts and ameloblasts. At this developmental stage, matrix vesicles (mv) are still intact and there is no overt sign of mineral deposition. Secretory granules (sg) in presecretory stage ameloblasts facing pulp show some immunoreactivity, indicating that the first amelogenin molecules originate, at least in part, from these cells.
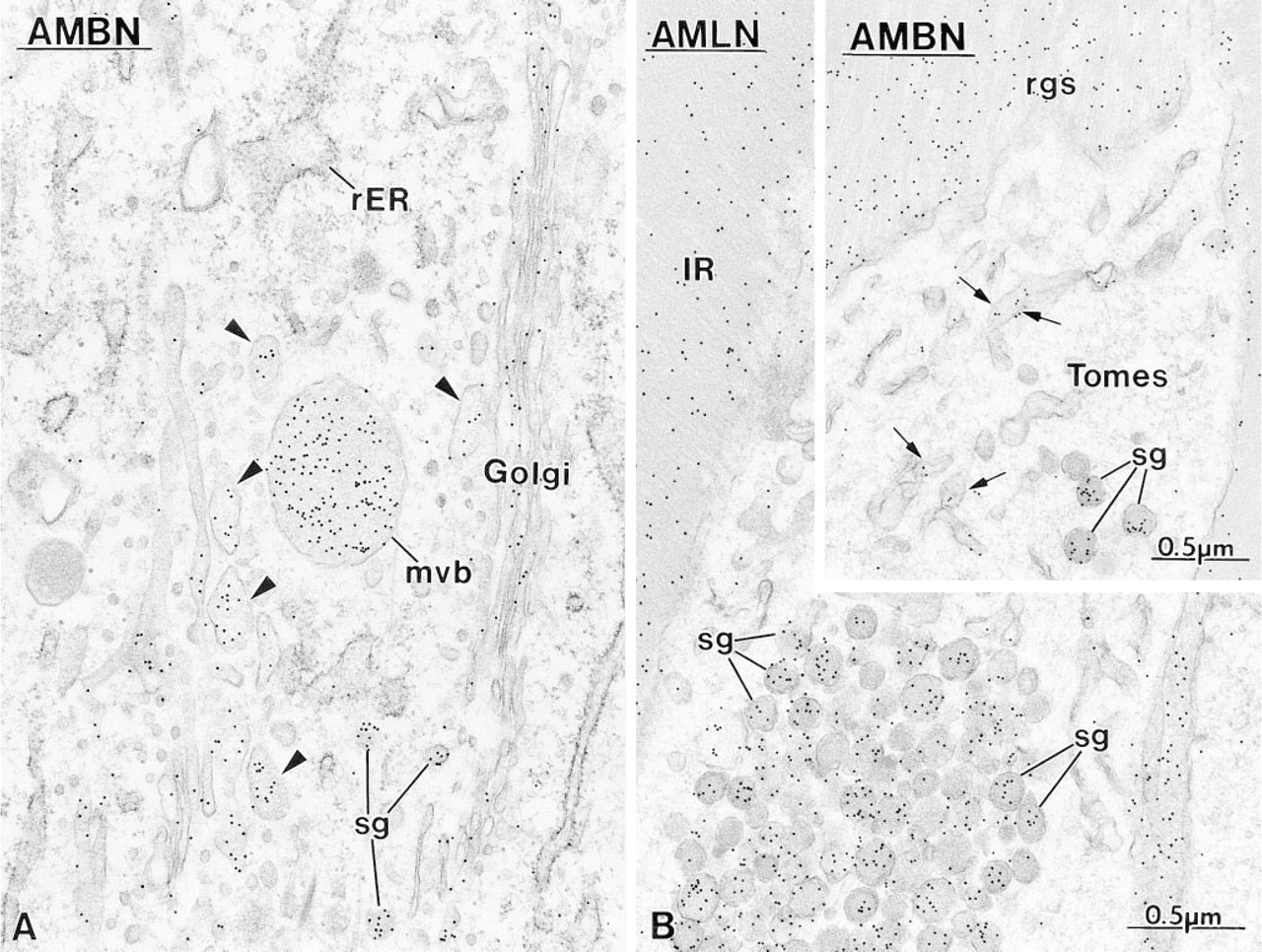
High-magnification micrographs illustrating the labeling obtained with anti-amelin (AMLN) and anti-ameloblastin (AMBN) antibodies over
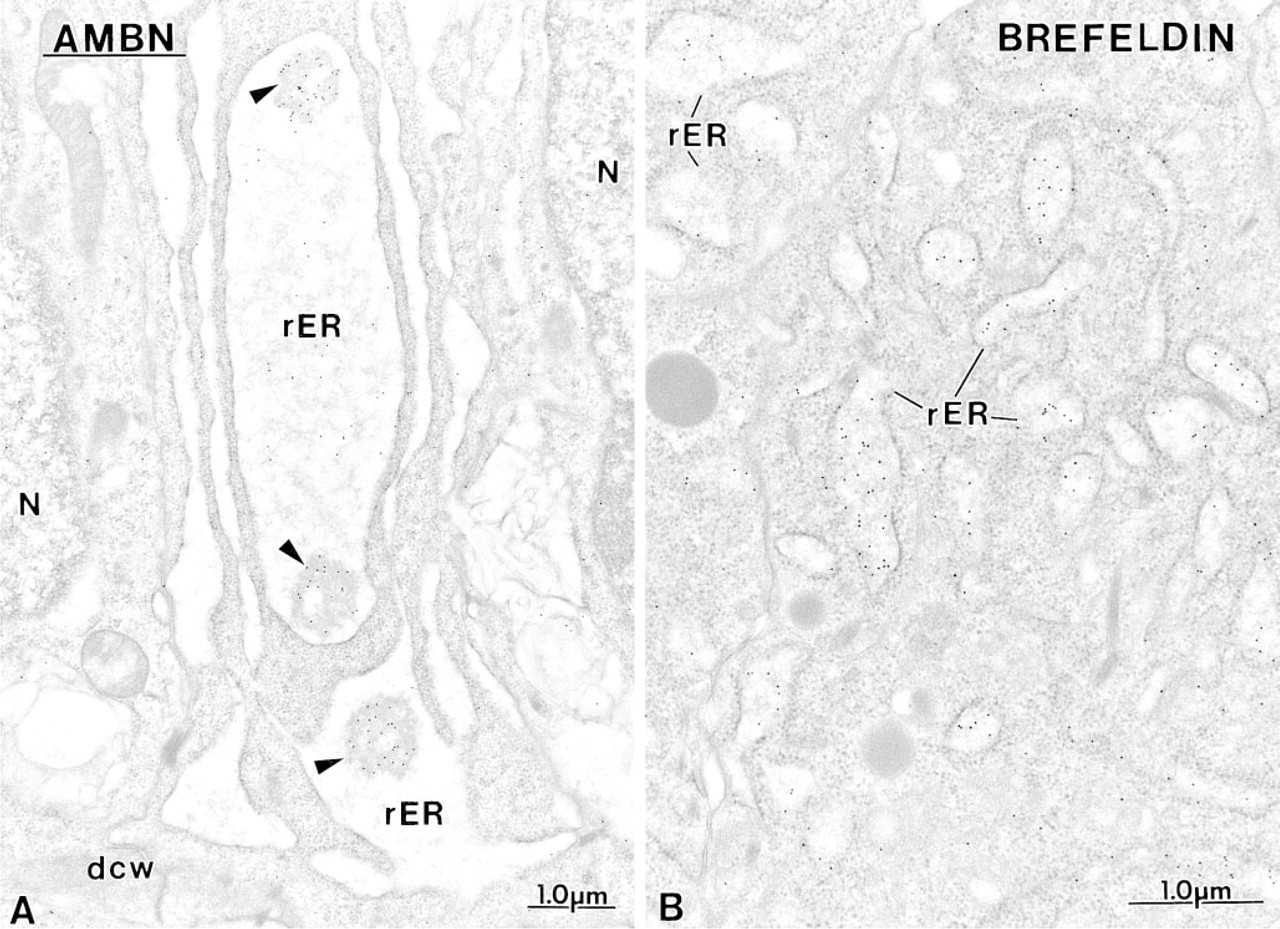
Immunocytochemical preparations illustrating the presence of ameloblastin (AMBN) in the rough endoplasmic reticulum (rER) of early
Immunolabeling for Enamel Proteins
Presecretory Stage. The labeling density over the Golgi apparatus and secretory granules increased with all three antibodies as enamel matrix accumulated extra-cellularly. However, in the early presecretory stage, when the basement membrane between ameleloblasts and odontoblasts was still intact, only the anti-amelo-genin antibody resulted in labeling over secretory granules (Figure 4A). Typically, dispersed labeling for amelogenin was seen over the forming extracellular matrix at the time of terminal differentiation of odon-toblasts before removal of the basement membrane (Figure 4A). Gold particles were associated with filaments of the lamina fibroreticularis (Figure 4A). Amelo-blastin was not immunodetected extracellularly at this early time with both the anti-amelin and the anti-ameloblastin antibody (Figures 4B and 4C), and appeared later over patches of electron-dense matrix in the mantle dentin. These patches labeled intensely for amelogenin (Figure 4D) but showed comparatively less immunoreactivity with anti-amelin (Figure 4E) and even less with anti-ameloblastin, despite the presence of gold particles over secretory granules (Figure 4F). The patches increased in number and size in an incisal direction and eventually were replaced by an initial layer of enamel showing crystal “ghosts.”
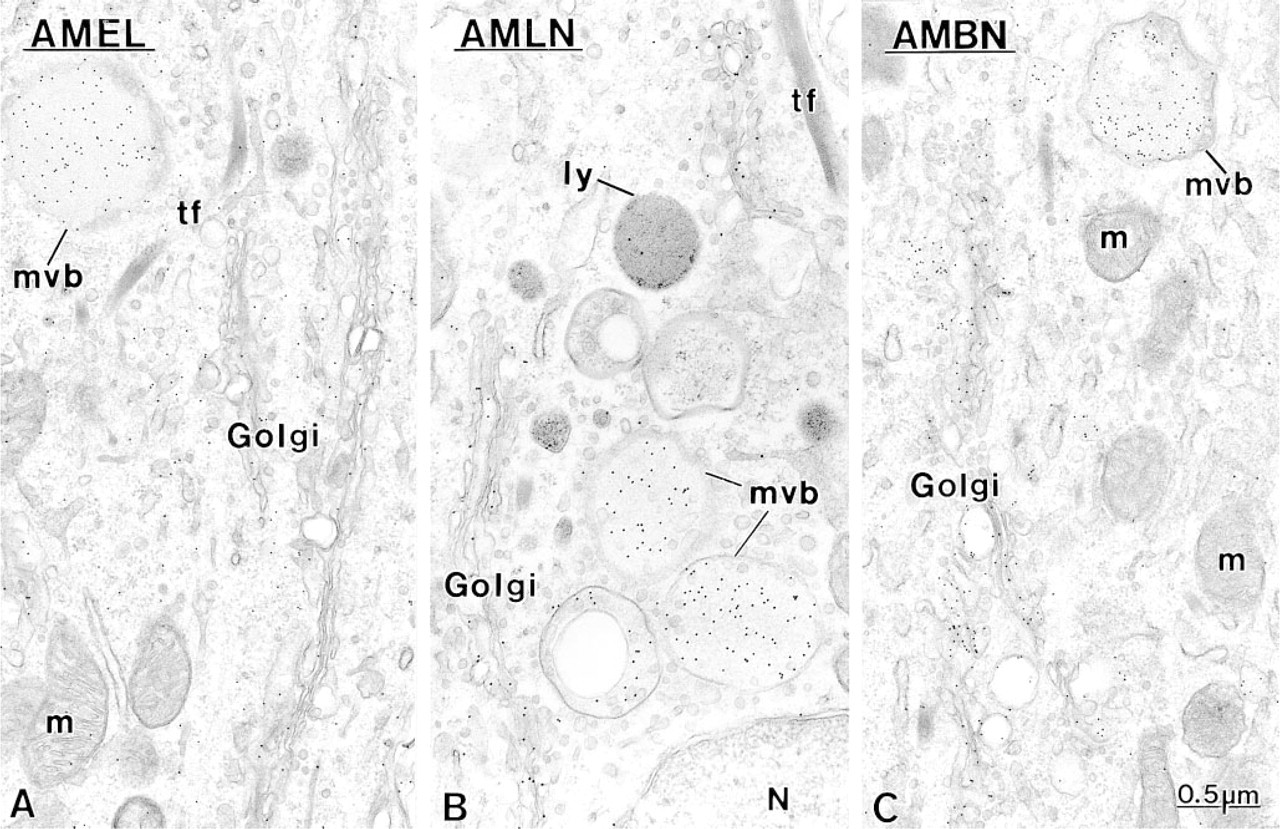
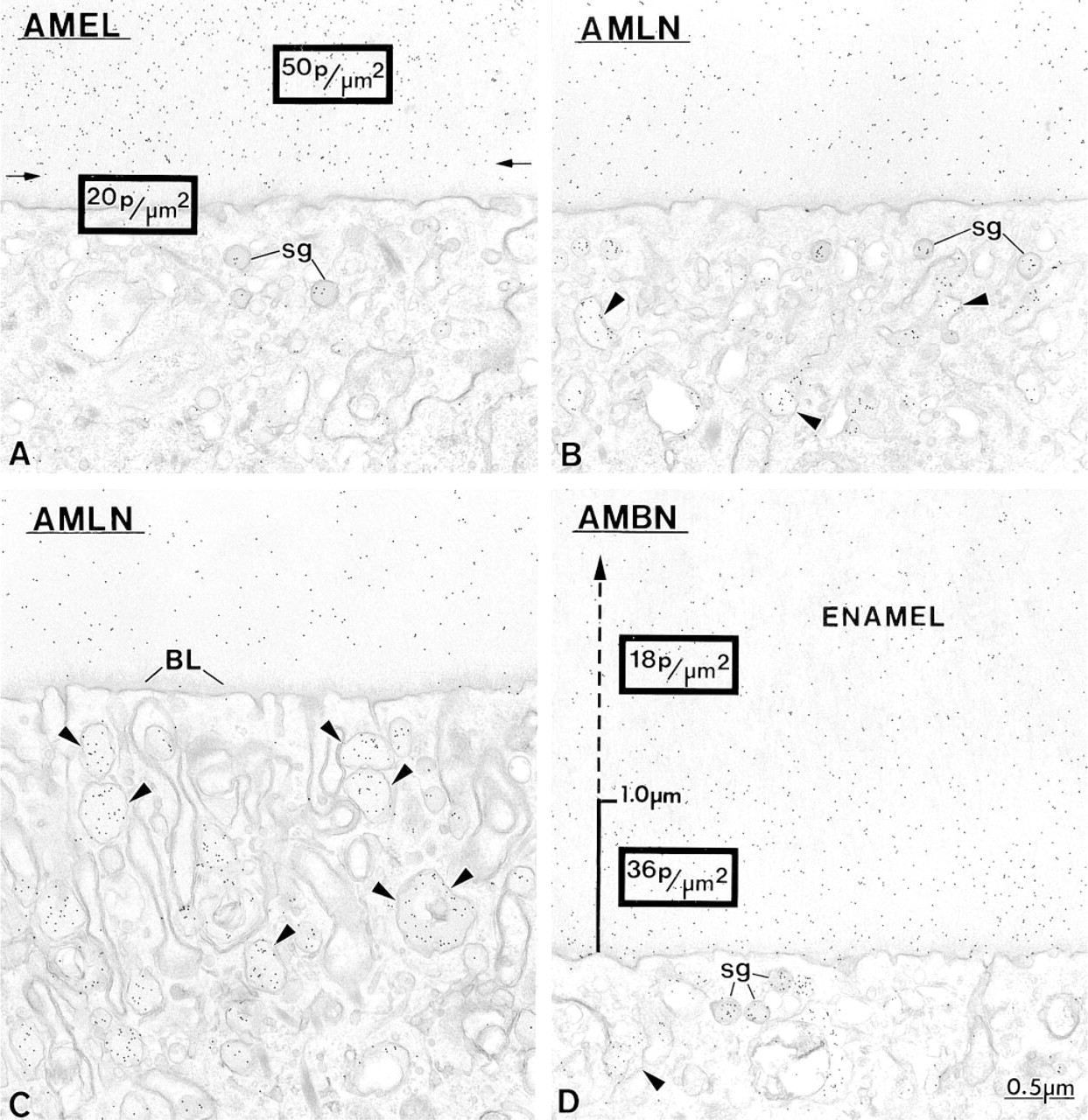
Secretory Stage. Immunoreactivity was most intense in secretory stage ameloblasts with all the antibodies. Many gold particles were present over the saccules of the Golgi apparatus, secretory granules in the Golgi region, and in Tomes' process, and over mutltivesicular bodies (Figure 5). Occasionally, labeling for amelo-blastin was present over smooth tubular elements in Tomes' processes (Figure 5 inset). Few or no gold particles were seen in association with the rough endo-plasmic reticulum in normal rats (Figure 5A). However, after treatment with brefeldin A, a fungal metabolite that blocks transport of protein from the rough endoplasmic reticulum to the Golgi apparatus (Lippincott-Schwartz et al. 1989), ameloblastin immunoreactivity was present over the rough endoplasmic reticulum, even in early presecretory stage ameloblasts in which protein synthetic organelles showed very little or no labeling under normal conditions (Figure 6). Initial enamel labeled differentially with the three antibodies. Anti-amelogenin showed gold particles throughout the entire enamel layer (not illustrated), whereas immunolabeling with anti-amelin and anti-ameloblastin antibodies was more intense at the forming enamel surface and weaker more internally (deeper locations) (Figure 7). The concentrated nature of immunolabeling at the enamel surface was particularly evident with the anti-ameloblastin antibody (Figure 7B). In some cases, amorphous patches of matrix were discernible between initial enamel and dentin, and these showed almost no immu-noreactivity with anti-ameloblastin (Figure 7B inset).
Immunocytochemical preparations with
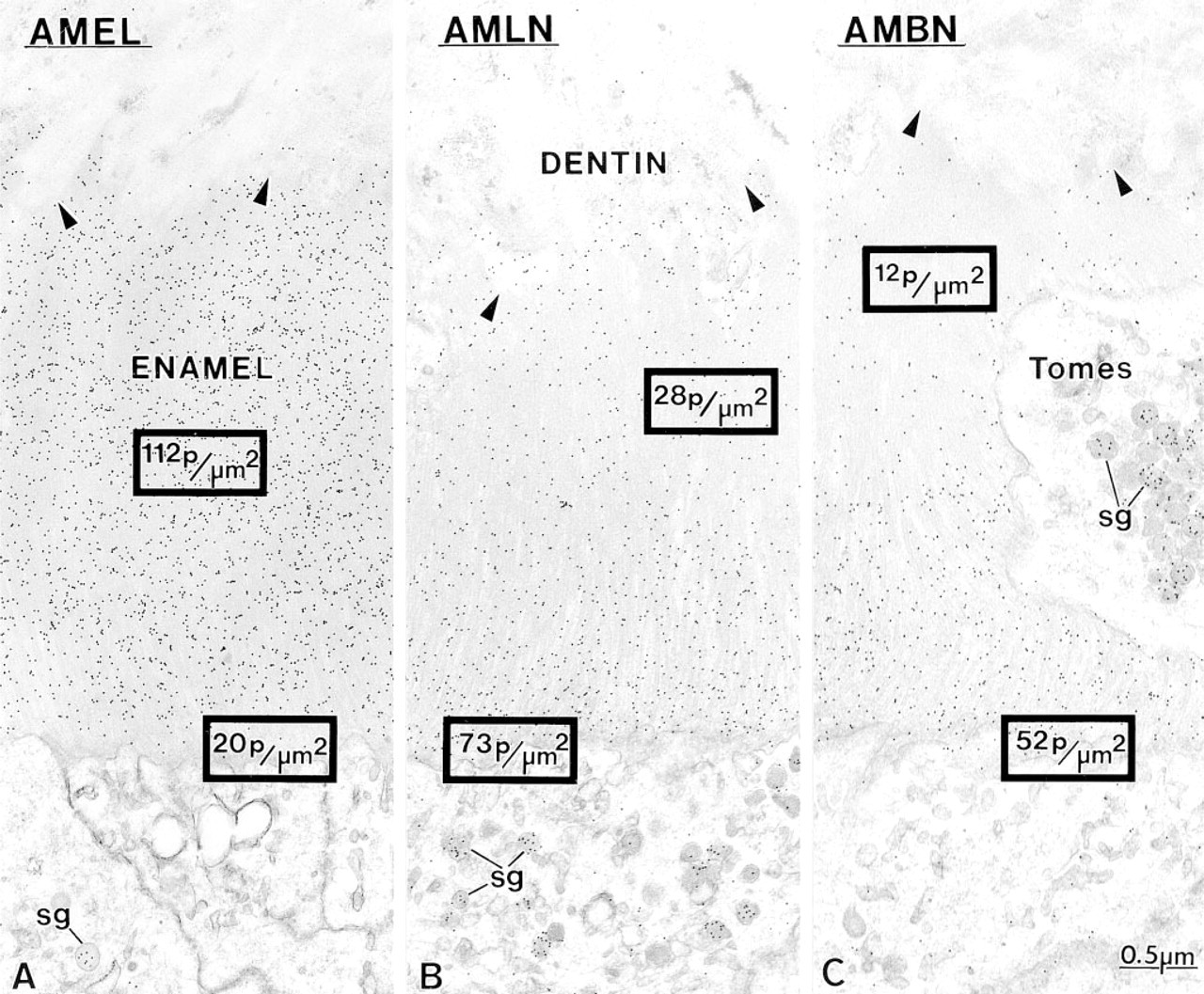
Anti-amelogenin and anti-amelin antibodies intensely labeled patches of organic matrix seen along the lateral surfaces of presecretory and secretory stage ameloblasts (Figures 4D, 4E, and 7A), but very few gold particles were seen over them with anti-ameloblastin (Figure 7B). In the early secretory stage (start of inner enamel), anti-amelogenin antibodies labeled the enamel intensely but showed a region of approximately 250 nm with less labeling at both the rod and interrod enamel growth sites (Figures 8A and 9). Immunolabeling with anti-amelin was seen throughout the enamel layer, but the reaction at growth sites was more intense than within deeper regions of the enamel (Figures 8B, 9, and 10A). Anti-ameloblastin antibodies clearly showed an inverse pattern compared to anti-amelogenin; many gold particles were present at growth sites and their density decreased relatively rapidly over a distance of ~1 μm to very few over the rest of the enamel layer (Figures 8C, 9, 10B, and 11A). The density of immunolabeling with anti-amelin was generally higher than with antiameloblastin, but in relative terms anti-ameloblastin showed the highest percentage of gold particles closest to the cell surface (Figure 9). The labeling at enamel growth sites obtained with anti-ameloblastin was noticeably reduced in rats treated with brefeldin A (Figure 11B) or cycloheximide (Figure 12).
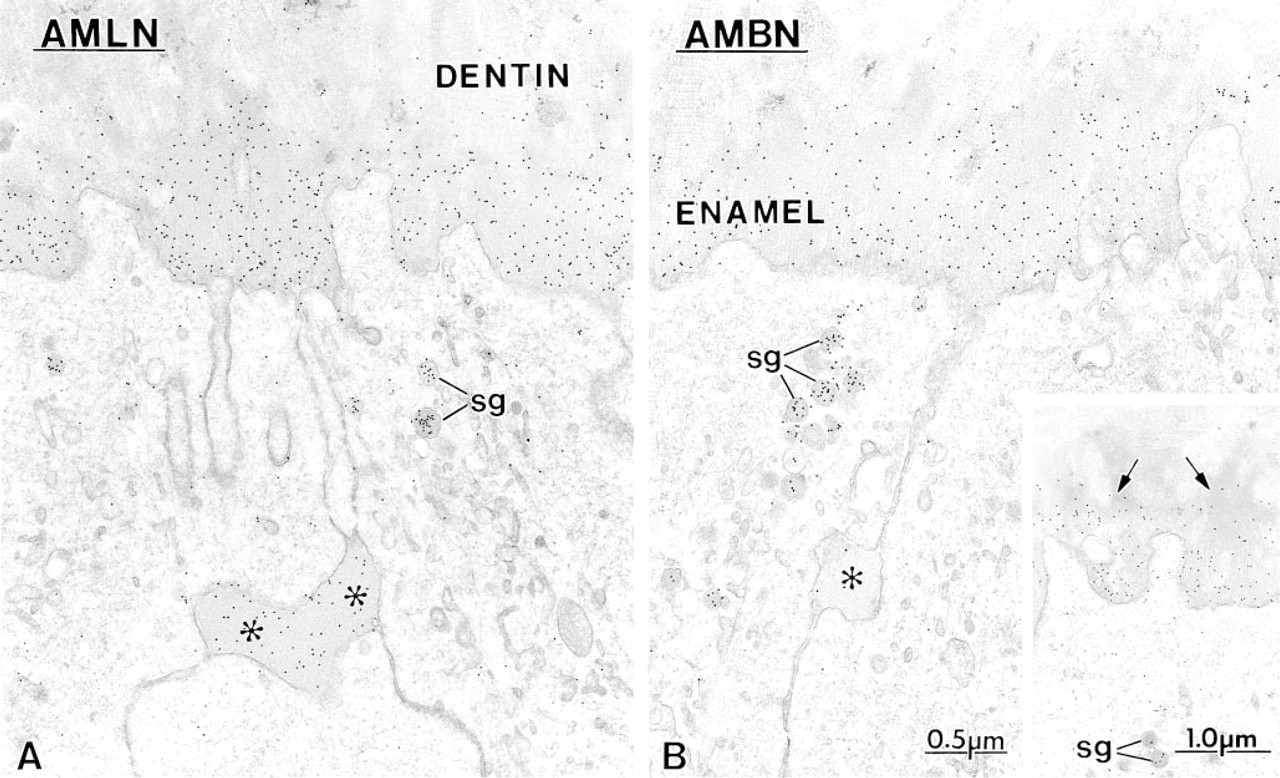
Micrographs of early inner enamel (secretory stage) when rods start to form, illustrating the interrod secretory surface of ameloblasts and associated enamel. Incubation with
Maturation Stage. During the early maturation stage, anti-amelogenin, anti-ameloblastin, and anti-amelin antibodies still showed immunoreactivity over the Golgi apparatus, secretory granules and endosomal/lysosomal structures of ameloblasts (Figures 13 and Figures 14). Granular and amorphous material located in the apical membrane infoldings of ruffle-ended amelo-blasts also showed the presence of gold particles (Figure 14C). As in the secretory stage, the antibodies yielded a differential immunolabeling over enamel (Figures 9 and Figures 14). Anti-amelogenin was immunoreactive throughout the enamel layer, except near the cell surface (Figures 9 and Figures 14A), whereas anti-amelin and anti-ameloblastin both showed a decreasing gradient away from the cell surface (Figures 9 and Figures 14D). At a later part of the maturation stage when rods are visible (Warshawsky and Smith 1974), the residual matrix appeared as a network of strands denser at the periphery of the interrod and rod profiles. Immunolabeling with anti-amelogenin and anti-amelin was still clearly visible at this time but was significantly reduced compared to early maturation (compare Figures 14A and 14B with 15A and 15B). Gold particles were associated with the matrix strands and appeared concentrated at the periphery of the rods (Figures 15A-15C). At this later time, anti-ameloblastin showed very weak immunolabeling at the enamel surface, but almost no gold particles were found over the matrix in deeper regions (Figure 15D). By mid- to late maturation, an electron-dense material accumulated in spaces near the dentino-enamel junction previously occupied by the extremity of the distal portion of Tomes' process; all three antibodies immunoreacted with this material (Figure 16). Anti-amelogenin and anti-amelin also labeled organic material found within or at the periphery of dentin tubules (Figure 17A inset).
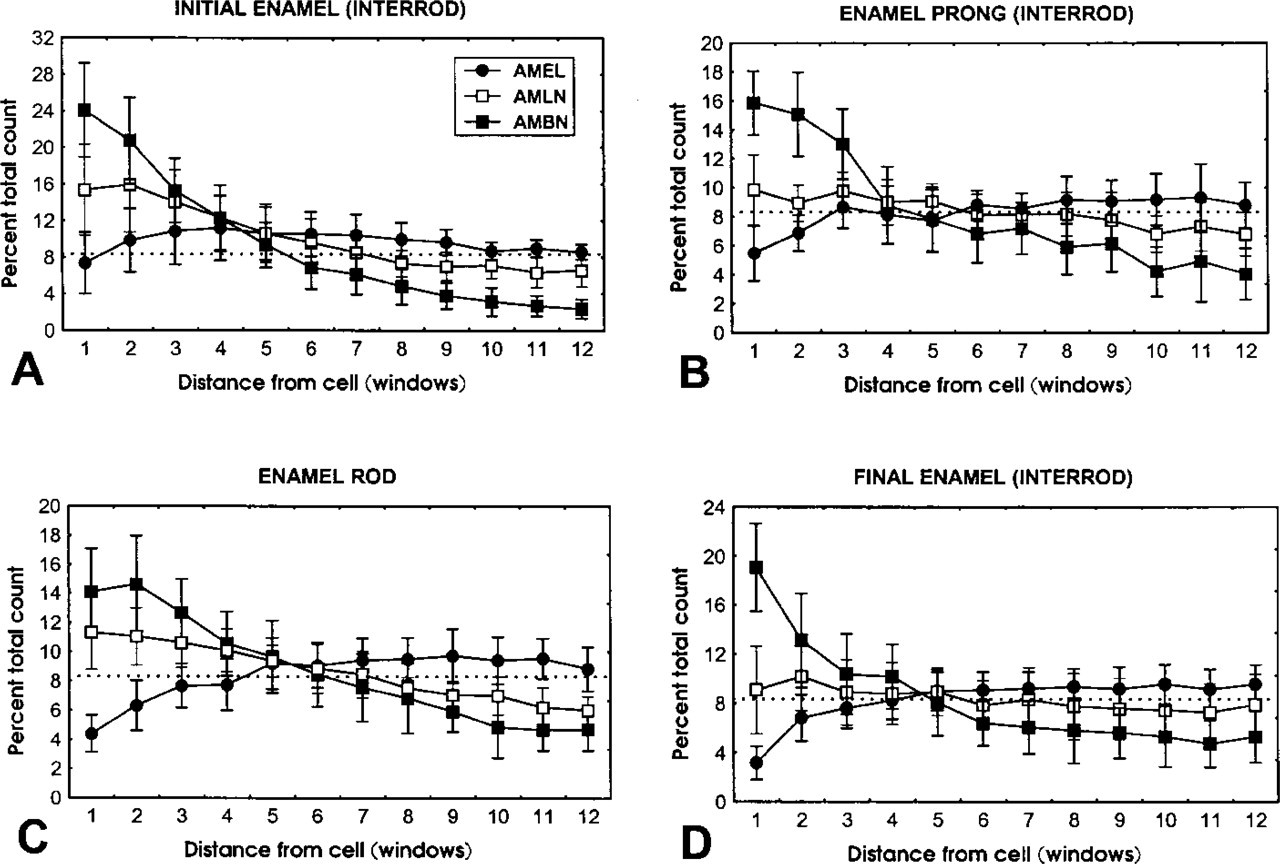
Categorization plots showing relative number of gold particles ± SD (expressed as percent of total particles counted per strip of 12 windows per antibody) obtained with anti-amelogenin (AMEL) (solid circles), anti-amelin (AMLN) (open square), and anti-ameloblastin (AMBN) (solid square) over
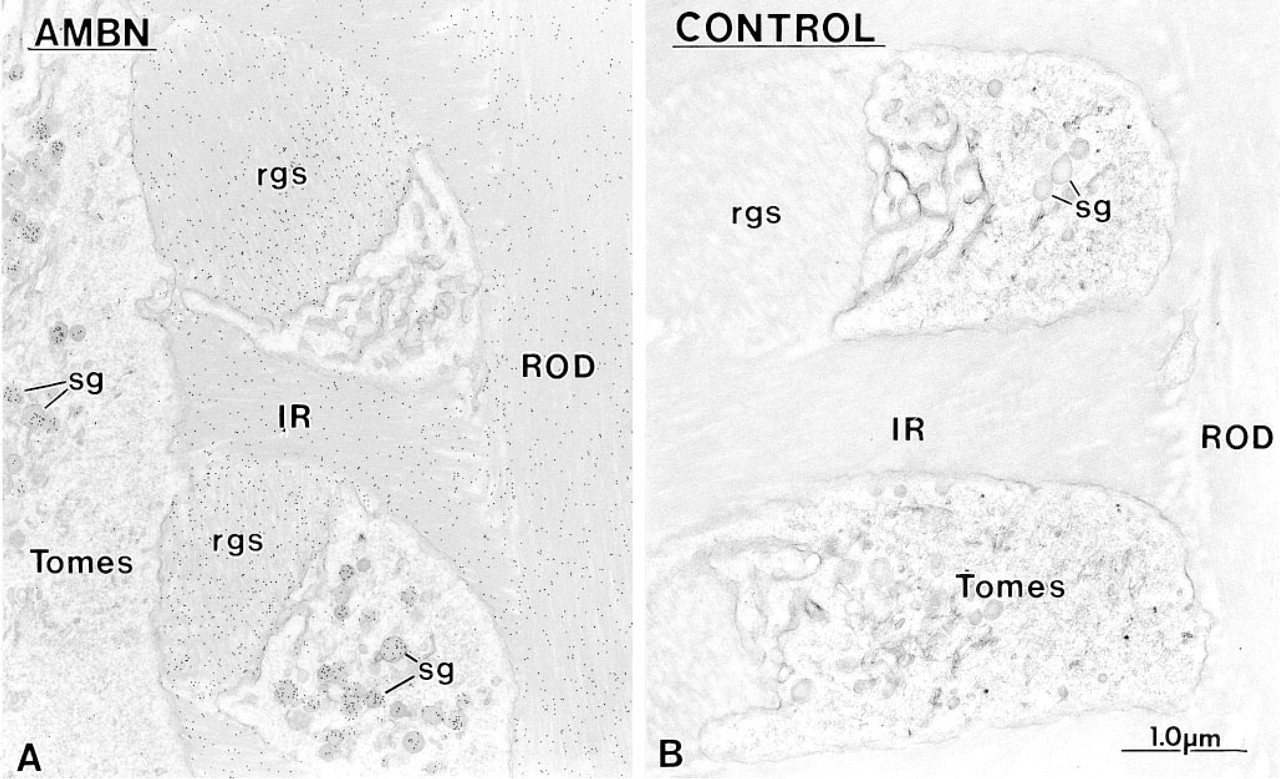
Immunocytochemical preparations illustrating the labeling obtained at rod growth sites with
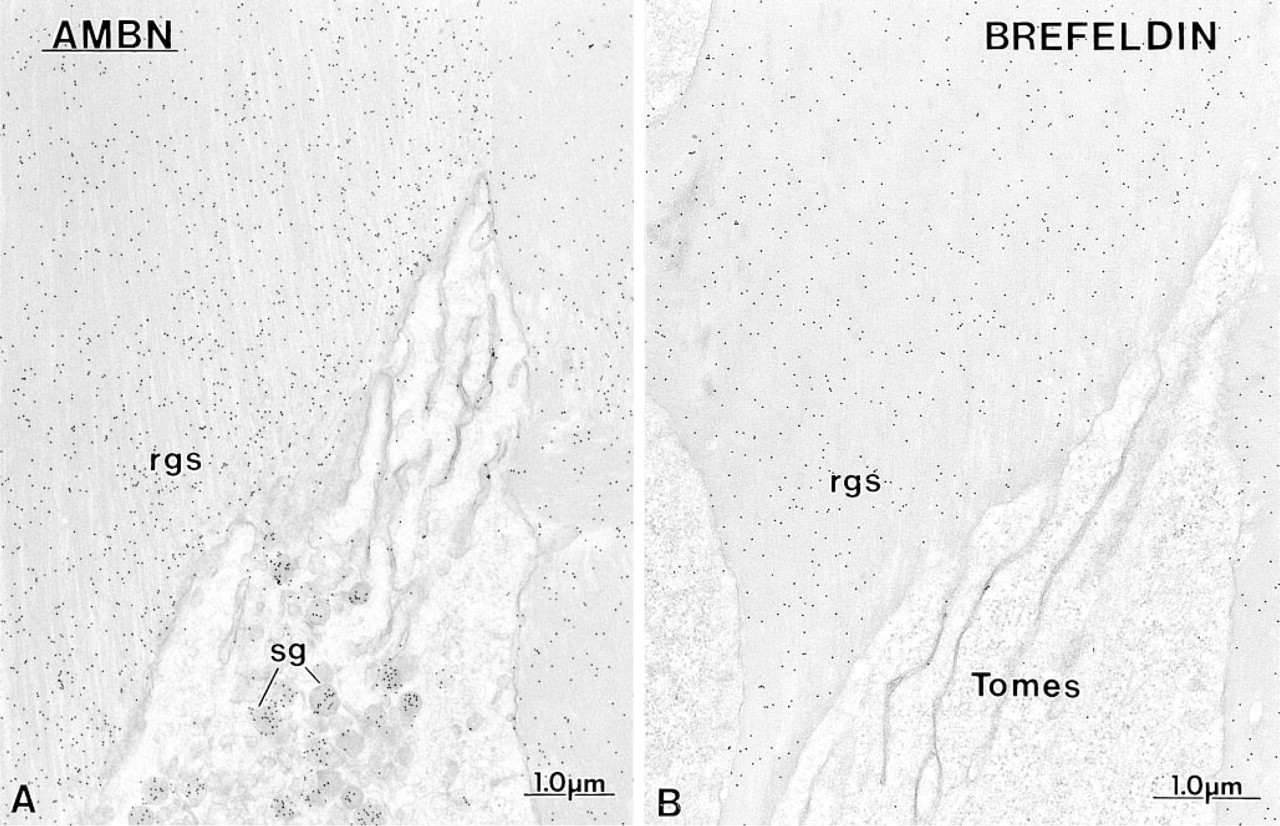
Comparative immunocytochemical preparations with anti-ameloblastin (AMBN) antibodies illustrating the labeling at the rod growth site (rgs) in
Controls
Control incubations with preimmune sera, rabbit antibodies to molecules unrelated to enamel proteins [e.g., immunoglobulins (Figure 10B)] or with protein A-gold alone (data not shown) revealed only a few randomly dispersed gold particles throughout the tissue section. The differential labeling obtained with the three anti-bodies and the reduction of anti-ameloblastin immunolabeling after cycloheximide treatment of animals also served as internal controls of the specificity of the binding, (i.e., nonspecific sticking would not be expected to generate the interrelated differential labelings described in Figure 9). In addition, anti-amelin, anti-ameloblastin, and anti-amelogenin showed a labeling similar to that obtained in control incubations over protein synthetic organelles of odontoblasts (Figure 17B) and osteoblasts and over dentin (Figure 17A) and bone, all of which are indicative of the tissue specificity of these antibodies.
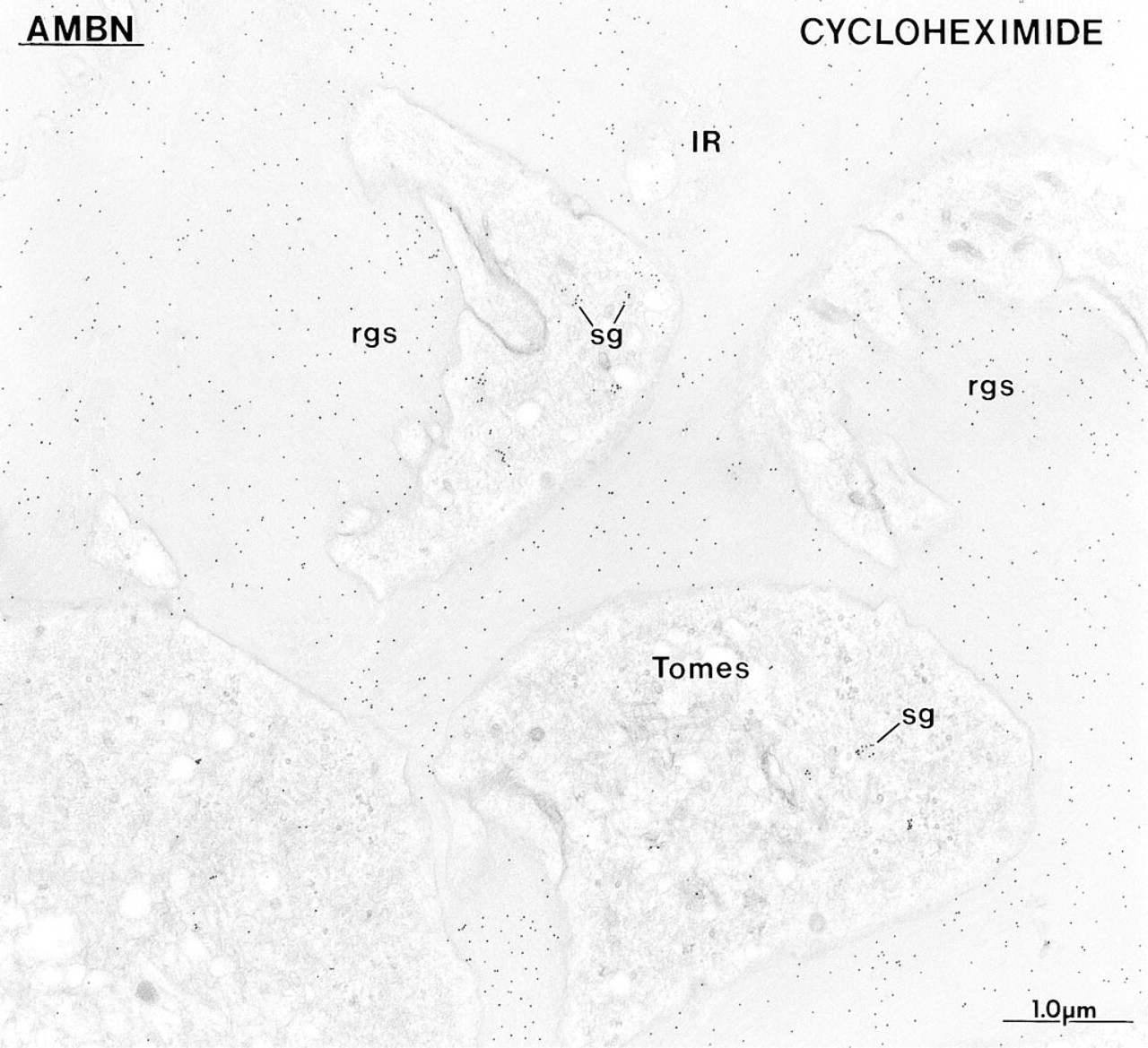
The concentration of immunolabeling at the rod growth site (rgs) observed with anti-ameloblastin (AMBN) antibodies in normal rats (see Figure 10A) is no longer apparent at 6 hr after cycloheximide administration to inhibit protein secretion, and the labeling density over enamel matrix at the rod growth sites is now similar to that over the surrounding interrod enamel (IR). The presence of a few secretory granules (sg) in some Tomes' processes indicates that by 6 hr ameloblasts start to recuperate from the drug.
Discussion
There is now clear evidence indicating that matrix proteins are differentially distributed across the enamel layer, but their interrelationships are still unknown. Immunoreactivity for a 32-kD porcine nonamelogenin (enamelin) is most intense near secretory surfaces of ameloblasts and that for 13-17-kD nonamelogenin (sheathlin) fragments and an 89-kD enamelin is preferentially found in the surface layer near Tomes' processes (Uchida et al. 1991a,b). Of these, 15-kD fragments localize along the nonsecretory surfaces of Tomes' processes (Uchida et al. 1995). Fragments of sheathlin also accumulate within the “sheath spaces” (Uchida et al. 1991a,b; Murakami et al. 1997). The degradation pattern of certain nonamelogenins (Uchida et al. 1991a,b; Murakami et al. 1997) and studies with protein secretion inhibitors (Nanci et al. 1996) indicate that proteins at enamel growth sites are short-lived and therefore probably play a transient role at that site. Lectin cytochemistry further shows that some of these proteins are glycosylated (Nanci et al. 1989,1996). High iron diamine staining for sulfated glycoconjugates results in preferential staining near secretory surfaces (Kogaya 1994) and immunoreactivity for phosphoserine has also been reported at sites at which enamel crystals grow in length (Nanci et al. 1996). The situation is more complex for amelogenins which, for a long time, have been assumed to form a thixotropic gel that allows free mixing of components and, hence, should lead to their homogeneous distribution (discussed in Smith et al. 1989; Smith 1998). Consistent with this theory, an early immunolocalization study of enamel proteins using a general anti-amelogenin antibody suggested a uniform distribution of matrix constituents (Nanci et al. 1985). However, a recent comparative analysis with a variety of anti-bodies unexpectedly revealed that some amelogenins and/or their degradation products, although enriched throughout the forming enamel layer, are present in lower amounts very near the plasma membrane of ameloblasts at which proteins are secreted (Nanci et al. 1996). Other studies have indicated that parental (25-kD) amelogenins are concentrated in the outer layer of immature enamel (Uchida et al. 1991b). The present results with an antibody to recombinant mouse amelogenin (M179) demonstrate quantitatively that amelogenin (intact molecule, isoforms, and/or processing products) is less concentrated near secretory surfaces, from the secretory to the early maturation stage.
Early maturation stage ameloblasts still show important immunolabeling with
The difference in immunolabeling with the two antibodies to ameloblastin reflects the antigen used to induce them. Anti-amelin was elicited using the whole-length recombinant molecule (Fong et al. 1996b). Therefore, it will recognize several epitopes along the entire molecule, albeit with variable avidity, such that the nascent protein, splice variants, and all fragments resulting from extracellular processing can be immunodetected. Anti-ameloblastin was generated against approximately the middle portion (residues 175-348) of the recombinant protein (Krebsbach et al. 1996) and it will react with the intact protein and fragments containing epitopes from this region. Fragments from the N-terminal 174 and C-terminal 74 amino acids are not recognized by the anti-ameloblastin antibody. The immunoblotting pattern with the two antibodies to ameloblastin is consistent with their expected binding and with what has been reported using synthetic peptide antibodies raised against various portions of the rat ameloblastin molecule (Uchida et al. 1997). It is noteworthy that the staining on blots from normal and cycloheximide-treated rats shows similarities with a 65-kD sulfated nonamelogenin (Smith et al. 1995; Smith and Nanci 1996). In addition, both antibodies to ameloblastin reveal in enamel organ cells some proteins near 65 kD across amelogenesis. No corresponding proteins are seen in maturation stage enamel samples, raising the possibility that they may not represent a secretory protein or that they turn over extremely rapidly after they are released extracellularly.
Immunocytochemical preparations with
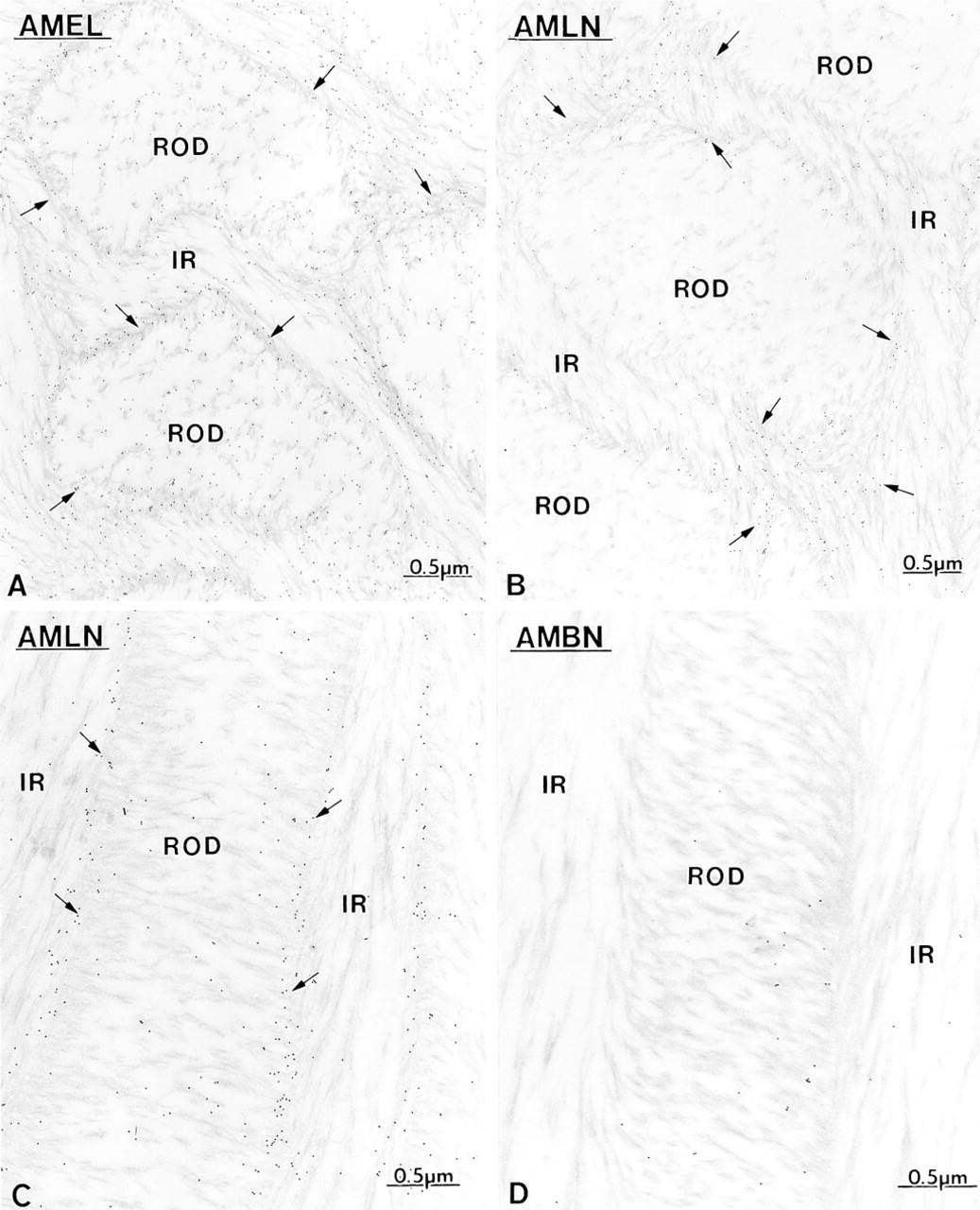
As enamel matures, rod profiles become visible and their periphery (arrows) is outlined by a denser organic matrix. This matrix is referred to as the “prism sheath” in higher mammals.
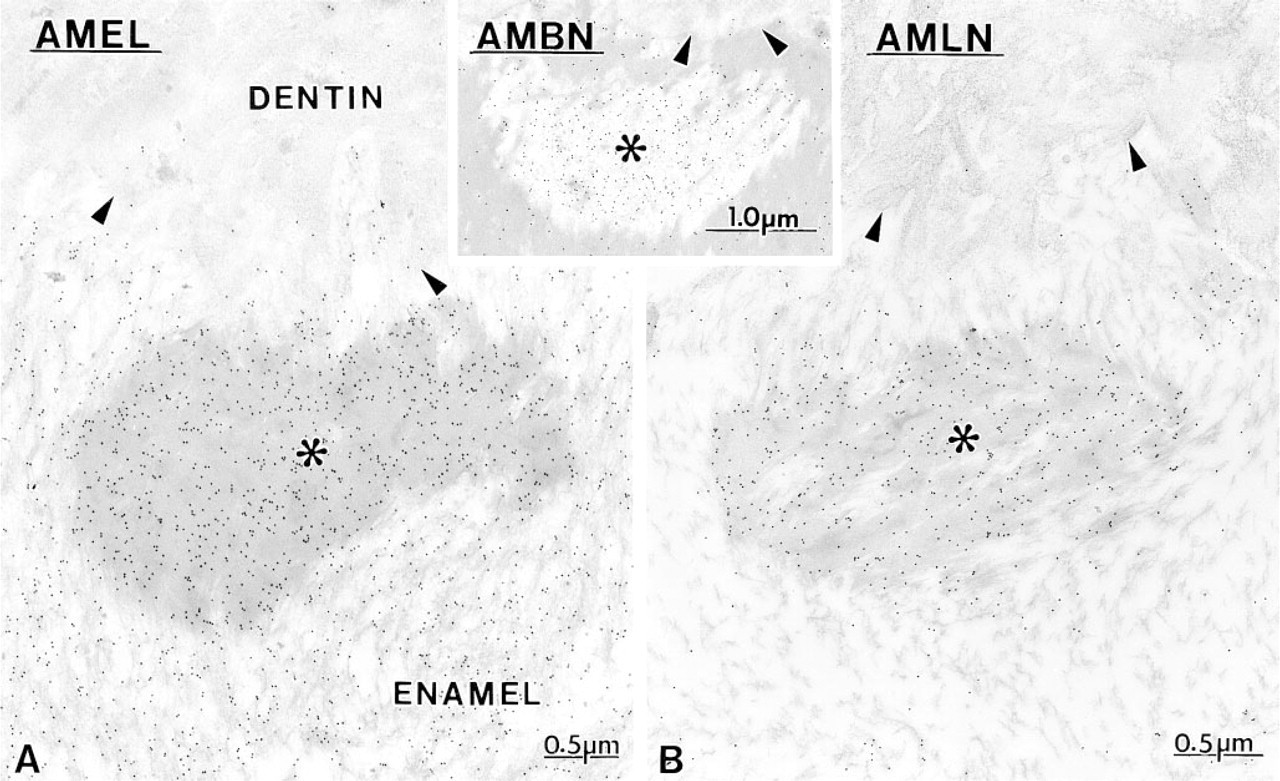
When inner enamel starts to form, there are spaces (asterisks) that appear near the dentino-enamel junction (arrowheads). (Inset) During the secretory stage and the beginning of the maturation stage, these spaces appear relatively empty and contain a fine granular material that immunoreacts with various antibodies to enamel proteins, such as anti-ameloblastin (AMBN).
The changes in immunolabeling patterns for amelo-blastin after inhibition of secretion with cycloheximide or brefeldin A demonstrate that this nonamelogenin accumulates transiently at enamel growth sites. This is particularly evident in normal rats with the anti-ameloblastin antibody, for which the differential immunolabeling across the enamel layer is most pronounced. There is a noticeable decline in immunoreactivity for ameloblastin at 1.25 mm away from the cell surface, a distance corresponding to about 2.2 hr of enamel appositional growth in rat incisors (Smith and Nanci 1989b). Unexpectedly, this position corresponds to the point at which the relative concentration of amelogenin stabilizes across the enamel layer (see Figure 9). Taken together, the immunoblotting and immunolabeling data demonstrate that the processing of ameloblastin is extremely rapid, as has been shown for the 65-kD sulfated protein in dynamic radiolabeling studies (Smith et al. 1995; Smith and Nanci 1996). Although the relationship between these two proteins remains to be determined, they appear to be at least similar in molecular weight and behavior. Our findings are also consistent with the proposal that ameloblastin undergoes an initial cleavage that generates a stable, small N-terminal fragment and a large C-terminal fragment, which then undergoes rapid degradation (Uchida et al. 1997). If the proteolytic processing rate of enamel proteins were equal to the rate of addition of new ones and no products leave enamel, one would expect that immunolabeling for the intact molecule and its fragments would eventually equilibrate, resulting in more or less uniform immunolabeling throughout the layer. The gradient observed by immunocytochemistry suggests that there exists some mechanism that favors the retention of nascent proteins for a short period of time near the area at which they are secreted and/or that some processing products are removed from the enamel layer as new molecules are added. However, because the rates of enamel protein deposition, processing, diffusion, and/or removal are presumably in a steady state during the active phase of enamel formation, the distribution of enamel proteins will appear to be static over time (see Figure 18 for schematic illustration).
The degree of labeling over
Conceptually, fragments of proteins will either persist and accumulate at lower molecular weights or they will leave the enamel layer. Smaller fragments (from several to few or single amino acids) could diffuse between ameloblasts out of the enamel organ or be endocytosed by cells. It is not possible to distinguish between these two scenarios from the data in this study. Some patches of matrix near the apical portions of ameloblasts show intense immunoreactivity with anti-amelin but very little with anti-ameloblastin antibodies. Considering the nature of the two antibodies, it is likely that the immunolabeling associated with these patches derives from the presence of N- and/or C-terminal fragments rather than intact ameloblastin. It should be noted that material along the basolateral aspects of ameloblasts also shows amelogenin immunoreactivity (discussed in Nanci and Smith 1992). In addition, this material is radiolabeled within less than 4 hr after injections of 3H amino acids, indicating that it contains at least some relatively young proteins (Nanci and Smith 1992). Therefore, enamel proteins found basolaterally may derive from ectopic secretion and/or diffusion of protein fragments away from the enamel layer. The presence of some low molecular weight proteins in ameloblasts and of immunolabeling within their endosomal/lysosomal elements with both antibodies to ameloblastin suggests that there is also active endocytosis from either the apical or the lateral surfaces of ameloblasts (reviewed in Nanci and Smith 1992). Such a possibility is consistent with the immunodetection of ameloblastin epitopes in coated vesicles (Uchida et al. 1997) and in smooth tubular elements in Tomes' processes (see Figure 7B inset).
Correlation of the in situ hybridization data with the biochemical and immunocytochemical results reaffirms the notion that enamel proteins are expressed at many times during amelogenesis (reviewed in Nanci and Smith 1992). Messenger RNAs for amelogenin and ameloblastin are both expressed at early times (Snead et al. 1988; Cerny et al. 1996; Fong et al. 1996a; Inage et al. 1996; Krebsbach et al. 1996; Lee et al. 1996). However, in situ hybridization does not permit unequivocally determination of which protein is expressed first when signals are very close, because it cannot be assumed that different probes will have similar degrees of penetration and efficiencies of interaction with the cellular RNA (features dependent on probe size and nucleotide ratios). In addition, the presence of an mRNA in a cell does not necessarily imply that it will sine qua non be translated into its corresponding protein. In the early presecretory stage, amelogenin is immunodetected extracellularly before ameloblastin, the latter being unequivocally identified only later when mineralization is about to start. The strong mRNA signals from both the ameloblastin and amelin probes at this early time contrast with their poor immunolabeling over the Golgi apparatus of ameloblasts. However, the detection of ameloblastin when cell products are “backed up” in the rough endoplasmic reticulum with brefeldin A clearly demonstrates that this nonamelogenin is indeed synthesized by presecretory stage ameloblasts. Therefore, it is either not secreted or it is made and released in amounts too small to be detected immunocytochemically. At this early developmental stage, it has also been reported, using amelin probes, that preodontoblasts transiently express both mRNA and protein signal for ameloblastin (Fong et al. 1998). As for preameloblasts, the lack of labeling in these other cells may be related to the immunodetectability threshold. It has also recently been shown that preameloblasts transiently express dentin sialo(phospho)protein, an odontoblast product (D'Souza et al. 1997; Ritchie et al. 1997). The expression of matrix proteins at early stages by cells that are not fully differentiated has important functional significance. In particular, the inverted expression pression of matrix proteins by epithelial and ectomes-enchymal cells as they differentiate, may be part of the reciprocal epithelio-mesenchymal signaling during tooth morphogenesis (D'Souza et al. 1997; Ritchie et al. 1997; Fong et al. 1998). In the maturation stage, message for amelogenin is found only during the early part of the stage, but that for ameloblastin persists throughout maturation. However, secreted proteins are not immunodetected, within the resolution limits of the approaches used, beyond the midmaturation stage. Therefore, the level of mRNA signal for ameloblastin does not necessarily correlate with the amount of protein actually synthesized and secreted by ameloblasts.
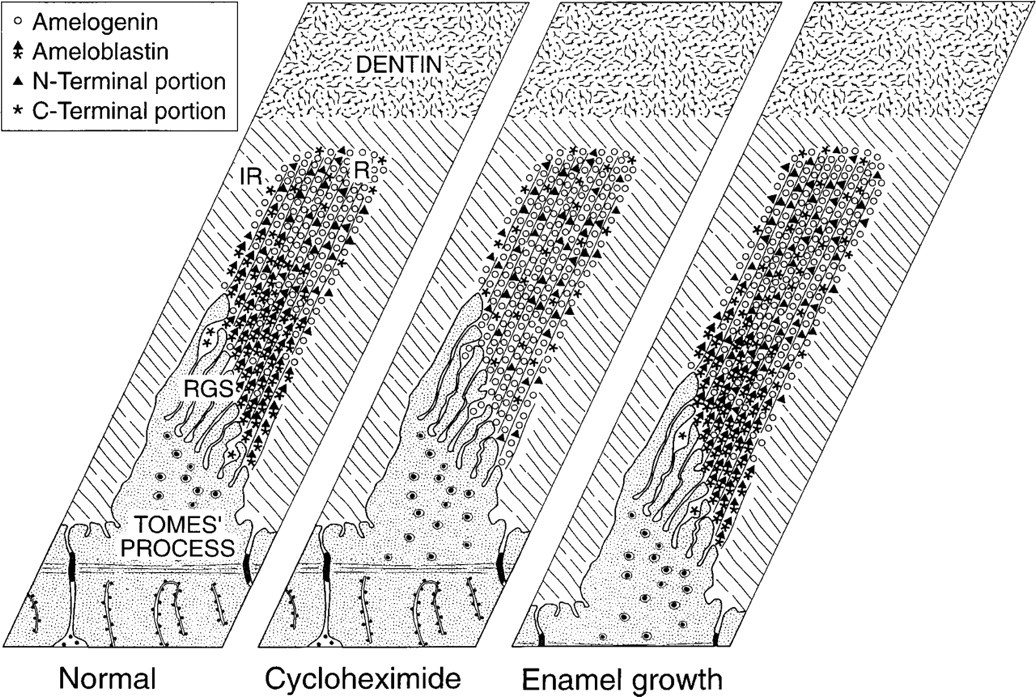
Schematic illustrations summarizing the distribution of amelogenin (○) and of intact and fragmented ameloblastin (∗▸) in the rod, as suggested by immunocytochemistry. In normal rats, amelogenin and ameloblastin show an inverse distribution pattern. There is less amelogenin immunoreactivity in a narrow region near the cell surface where proteins are secreted, whereas intact ameloblastin is more concentrated at the rod growth site. The N-terminal portion (▸) of the molecule persists for some time in deeper enamel, whereas most of fragments from the C-terminal portion (∗) leave the enamel. When protein secretion is arrested by cycloheximide treatment, the differential distribution of amelogenin and ameloblastin is no longer apparent. Amelogenin is now uniformly distributed, whereas there is only weak and diffuse labeling for ameloblastin throughout the enamel rod. Therefore, the newly secreted ameloblastin that piles up at enamel growth sites turns over rapidly. The right hand panel depicts what happens to the distribution of ameloblastin as the enamel layer thickens. Because the rates of enamel protein deposition, processing, diffusion, and/or removal are presumably in a steady state during the active phase of enamel formation, the thickness of enamel increases but the distribution of enamel proteins appears to remain static. IR, interrod enamel; R, rod enamel; RGS, rod growth site.
The early secretion of amelogenin at a time when odontoblasts have not yet fully differentiated, mantle predentin is not yet discernible, and enamel mineralization has not yet started suggests that this protein is multifunctional. Initially, it may participate in epithelial-mesenchymal events (Nanci and Smith 1992; Sawada and Nanci 1995) and, perhaps, crystal nucleation. However, because there is no overt sign of mineral deposition (e.g., crystals or their ghosts) among the initial patches of enamel proteins, it is likely that any role amelogenin may have in crystal nucleation is associated with the temporal expression of specific isoforms, extracellular processing of major isoforms, and/or the arrival of other proteins, such as ameloblastin. When enamel mineralization is ongoing, amelogenin may function to regulate growth in width and thickness of crystals (reviewed in Robinson et al. 1995; Simmer and Fincham 1995). This is not a unique circumstance; despite some uncertainty about the tissue specificity of tuftelin (Diekwisch et al. 1997; Zeichner-David et al. 1997), its early expression several days before mineralization starts has led to the suggestion that it may first have a role in cell signaling and then in mineral deposition (Zeichner-David et al. 1997). Noncollagenous bone/cementum proteins, such as bone sialoprotein and osteopontin, also have been associated with a variety of cellular and extracellular matrix activities (reviewed in Boskey 1995; Sodek et al. 1992; Denhardt and Guo 1993). Of note, it has recently been reported that deposition of these two typical noncollagenous proteins can occur in association with inner enamel epithelial cells (Bosshardt and Nanci 1997,1998). In contrast, the pattern of expression of ameloblastin suggests that it is more selective in its function. It is unequivocally immunodetected extracellularly only when mineralization is about to start. Subsequently, it associates preferentially with areas at which crystals elongate, strongly supporting the participation of this short-lived protein in events related to initiation and regulation of crystal elongation. The presence of little intact amelo-blastin (as judged by the weak labeling obtained with antibody against the midportion of the molecule) in patches of organic matrix, found in the forming mantle dentin and basolaterally between some ameloblasts, could in part explain why no evidence of mineral deposition is seen at these sites, and lends support to their proposed role in promoting mineralization. It also highlights the fact that extracellular processing starts as soon as secretion begins and, consequently, proteinases responsible for their degradation must be produced sooner than expected, during early amelogenesis.
Some nonamelogenins are believed to persist after removal of amelogenins during maturation (reviewed in Robinson et al. 1995). Of these, “tuft proteins” are found at the dentino-enamel junction (Robinson et al. 1975), whereas sheathlin is believed to accumulate in the so-called “prism sheath” (Uchida et al. 1995; Hu et al. 1997a). Patches of electron-dense organic matrix are clearly visible near the dentino-enamel junction from midmaturation and beyond. The spaces occupied by this matrix correspond to the position at which the distal portion of Tomes' process starts forming the enamel rod (Nylen et al. 1970). As the enamel layer thickens, Tomes' process is compressed by the enlarging rod and is eventually obliterated, leaving a space at its distal extremity between interrod and rod enamel (Warshawsky et al. 1981). During the secretory stage in the rat, there is relatively little organic matrix in these spaces and they generally appear as electron-lucent regions containing some fine granular material. However, as maturation proceeds and massive protein degradation takes place, the spaces become filled with electron-dense material (see Figure 16). Although these accumulations of matrix are situated at the dentino-enamel junction like “tuft protein” (Robinson et al. 1975; Robinson et al. 1989), they do not contain a single and distinct protein but consist essentially of a mixture of proteins, comprising at least intact amelogenin and/or its fragments and portions of ameloblastin.
In the region at which enamel rods are visible (Warshawsky and Smith 1974), there is a concentration of immunolabeling with anti-amelogenin and anti-amelin antibodies at the periphery of interrod and rod enamel. The comparatively higher density of immunolabeling at these sites does not represent a true concentration of protein as is found at enamel growth sites. It probably reflects the fact that, as a result of the maturation process, there remains more organic matrix at the periphery of interrod and rod enamel than within their bulk. Unlike the matrix patches at the dentino-enamel junction, the paucity of immunolabeling with the anti-ameloblastin antibody at the periphery of rods at later stages establishes that the antigenicity observed there is due to the presence of fragments rather than intact molecules. These fragments may passively accumulate in regions where there are spaces as they are displaced from the growing crystal environment and leave the enamel layer. Similarly, enamel proteins may passively diffuse into dentin tubules opposite maturing enamel.
In conclusion, we have shown that amelogenins and ameloblastin are differentially distributed throughout the forming enamel layer. During the appositional growth phase, enamel crystals elongate to their full length, whereas growth in width and thickness is contained (Robinson et al. 1995; Simmer and Fincham 1995; Smith 1998). Indeed, these two processes are spatially and temporally separated in the sense that crystal elongation is restricted to the secretory stage, whereas growth in width and thickness is limited to deeper regions of enamel during the appositional phase and then occurs predominantly during the maturation stage when amelogenins are removed (reviewed in Smith 1998). Because elongation is a continuous process, it would be expected that “fresh” matrix constituents are required to promote/regulate the addition of new mineral, whereas older ones are degraded or incorporated into the matrix. The combination of approaches we have used has provided some insights into the dynamics of proteins during enamel formation. In the case of ameloblastin, the intact protein accumulates preferentially at growth sites, where it likely plays a role in crystal promotion/elongation, and is then rapidly processed to lower molecular weight species. Amelogenins are more stable and generally attain their highest concentration a short distance away from the cell surface. These matrix events take place over a period of about 2 hr. The data further suggest that there may be an “inverse” link between the lesser concentration of amelogenins and the accumulation of ameloblastin at enamel growth sites. Amelogenins have been proposed to regulate crystal growth in volume (reviewed in Simmer and Fincham 1995), and it therefore may not be desirable to have high concentrations of such proteins possessing inhibitory potential at sites where crystals actively expand in length. A major question is, “what are the events or biophysical characteristics of amelogenin and/or ameloblastin that lead to their particular distribution?” Although self-assembly of certain proteins such as amelogenin may be important in defining their function (Moradian-Oldak et al. 1995; Paine et al. 1996; Paine and Snead 1997), it appears that the lack of interaction between amelogenins and some nonamelogenins may be a key factor leading to their differential distribution at growth sites and enabling them to differentially express their activity. The rapid extracellular processing of a nonamelogenin such as ameloblastin may also contribute to defining its activity by generating a number of protein fragments with different functional properties from those of the parent molecule.
Footnotes
Acknowledgements
Supported by a grant from the Medical Research Council of Canada to AN. PL was the recipient of a Burroughs-Wellcome studentship from the Medical Research Council of Canada.
We are grateful to Micheline Fortin for her assistance with tissue processing, sectioning, and quantification of the immunolabeling.