
Abstract
We describe a pre-embedding immunocytochemical method for visualization of the lysosomal enzyme cathepsin D in cultured cells. The protein was demonstrated at both light and electron microscopic levels by neutral-pH silver enhancement of ultrasmall (0.8-nm) gold particles conjugated to the antibodies. The best morphological preservation and the highest labeling density were achieved by initial fixation for 20 min at 4C in 4% paraformaldehyde (PFA) and 0.05% glutaraldehyde (GA) in 0.15 M sodium cacodylate buffer, followed by permeabilization in sodium borohydride. Three cell types were used: human foreskin fibroblasts, histocytic lymphoma (J-774) cells, and primary rat heart myocytes. In all three, cathepsin D was demonstrated in lysosome-like structures. The rat heart myocytes were also exposed to the redox cycling substance naphthazarin (5,8-dihydroxy-1,4-naph-thoquinone) to induce oxidative stress. This was done for such a short period of time that the cells initially did not show any signs of morphological damage and retained normal plasma membrane stability, although an early and clear redistribution of cathepsin D from membrane-bound structures to the cytosol was apparent. This redistribution was followed by cell degeneration and, eventually, by cell death.
I
Cathepsin D is a lysosomal aspartic protease that is present in almost all animal cells (Yamamoto 1995). It degrades proteins at low pH and exerts its activity inside lysosomal structures. Exposing enriched fractions of lysosomes to oxygen-derived free radicals has been found to cause leakage of enzymes from these organelles (Mak et al. 1983; Zdolsek and Svensson 1993). Moreover, Altona et al. (1984) found that anoxia-reoxy-genation injury of rat heart myocytes induced release of the lysosomal enzymes N-acetyl-β-
We propose that, during oxidative stress, increased amounts of hydrogen peroxide are produced that can diffuse freely and may therefore cross the lysosomal membrane and spread within the acidic vacuolar apparatus. Inside this compartment, the acidic milieu and the occurrence of reducing compounds (e.g., cysteine) would promote iron reduction and Fenton-like chemistry. This would give rise to hydroxyl radicals that could destabilize the lysosomal membranes (e.g., through peroxidation) and thereby cause leakage of lysosomal contents to the cytosol (Brunk et al. 1995a).
To study the actual localization of lysosomal enzymes in normal cells and in cells exposed to oxidative stress, we developed a pre-embedding method to visualize the lysosomal enzyme cathepsin D. This was done using ultrasmall gold particles and silver enhancement. To show the applicability of the method, three different types of cultured cells were used: human foreskin fibroblasts, an established line of histocytic lymphoma (J-774) cells, and primary rat heart myocytes. The labeling density and morphological preservation at different concentrations of paraformaldehyde (PFA) and glutaraldehyde (GA) were evaluated. Using TEM (transmission electron microscopy), we also found that cathepsin D in myocyte lysosomes was partially released into the cytosol during exposure to oxidative stress and that only later did cell degeneration and death occur.
Material and Methods
Chemicals
Glutamine, penicillin-G, streptomycin, Eagle's minimal essential medium (EMEM), Ham's F-10 medium, fetal calf serum, and dialyzed calf serum were purchased from GIBCO (Paisley, UK). Hydroquinone was obtained from BDH Ltd (Poole, UK); silver lactate, paraformaldehyde, and Epon-812 from Fluka AG (Buchs, Switzerland); and glutaraldehyde (vacuum distilled) from Agar Scientific (Essex, UK). Naphthazarin (5,8-dihydroxy-1,4-naphthoquinone) came from Aldrich Chemie (Steinheim, Germany). Collagen (Type I), sodium borohydride, sodium cacodylate, citric acid, and sodium citrate were supplied by Sigma (St Louis, MO). Gelatin (coldwater fish skin gelatin) and goat anti-rabbit IgG tagged with 0.8-nm gold particles were bought from Aurion (Wageningen, The Netherlands). Cathepsin D antibodies and the normal rabbit immunoglobulin fraction were from Dakopatts (Älvsjö, Sweden).
Cells and Culture Conditions
Human foreskin fibroblasts, AG-1518 (passages 15–25), were cultured in EMEM supplemented with 2 mM glutamine, 50 IU/ml penicillin-G, 50 μg/ml streptomycin, and 10% fetal calf serum. The established murine histocytic lymphoma cell line (J-774 cells) was cultured in Ham's F-10 medium supplemented with 10% fetal calf serum, 2 mM glutamine, 50 IU/ ml penicillin-G, and 50 μg/ml streptomycin.
These two cell lines were incubated in humidified air with 5% CO2 at 37C and were subcultured once a week. Before an experiment (24 hr), the cells were trypsinized (fibroblasts) or scraped (J-774 cells) and seeded into 35-mm Petri dishes (Costar; Cambridge, MA) at a density of 10,000 and 30,000 cells/cm2 for fibroblasts and J-774 cells, respectively. For LM (light microscopy), round glass coverslips (diameter 22 mm) were placed in the culture dishes before cell plating.
Heart myocytes were prepared from 2- to 3-day-old male and female Sprague-Dawley rats as previously described (Öllinger and Brunmark 1994). Cells were plated at a density of 50,000 cells/cm2 in culture dishes or on coverslips previously coated with 5.0 μg of Type I collagen/cm2 and then cultured in humidified air with 5% CO2 at 37C for 5–6 days before use in experiments. The culture medium, which consisted of EMEM supplemented with 10% dialyzed calf serum, 2 mM glutamine, 100 IU/ml penicillin-G, 100 μg/ml streptomycin, and 10 μg/ml cytosine-β-D-arabinofuranoside, was changed every second day.
Exposure to Naphthazarin
Myocytes were exposed to naphthazarin dissolved in 99% ethanol added in pre-warmed (37°C) Hank's balanced saline solution, pH 7.4 (final ethanol concentration 0.25%) and incubated at 37°C. Viability (i.e., plasma membrane integrity) was studied by analysis of lactate dehydrogenase (LDH) leakage, as described by Vassault (1985). LDH activity of the cells and in the surrounding medium was analyzed separately, and the viability was expressed as percentage of cell activity or of total LDH activity.
Immunogold Labeling
The cultures were fixed in different fixatives consisting of 2% or 4% PFA and between 0.05% and 1% GA in 0.15 M sodium cacodylate buffer, pH 7.6, for 20 min at 4C. The cells were washed in PBS and then immersed in a freshly prepared solution of sodium borohydride (0.01% or 0.05%) and 0.1% glycine in PBS, pH 7.4, for 10 min at room temperature (RT). Before and after each immunoincubation, cells were rinsed for at least 1 hr in PBS containing 20 mM NaN3, 0.8% BSA (bovine serum albumin), and 0.1% gelatin. This solution was also used to dilute primary and secondary antibodies. The cells were incubated in 1:100 diluted polyclonal rabbit cathepsin D antibodies overnight at 4C. After rinsing for 5 hr, the cells were incubated in 1:100 diluted goat anti-rabbit IgG or F(ab') tagged with 0.8-nm gold particles overnight at 4C. The cells were then rinsed for 1 hr in PBS containing 20 mM NaN3, 0.8% BSA, and 0.1% gelatin, and for 1 hr in PBS, fixed for 10 min in 2% glutaraldehyde in PBS, and finally washed in PBS at RT. Control cells in which the specific polyclonal antibody had been replaced (with immunoglobulin fraction of nonimmune rabbit serum or PBS containing 20 mM NaN3, 0.8% BSA and 0.1% gelatin) remained unstained.
Silver Enhancement Procedure
The cultures were rinsed three times with 20 mM HEPES buffer containing 280 mM sucrose (pH 6.8) and were then developed for 6 min. The developer solution was prepared by combining 6 parts gum arabic (500 g/liter in deionized water), 1 part HEPES buffer stock (200 mM HEPES; pH adjusted to 6.8 with NaOH), 1.5 parts hydroquinone (0.52 M in deionized water), and 1.5 parts silver lactate (37 mM in deionized water). The silver lactate was protected from light and was added just before use. The cultures were incubated with developer in a dark chamber at 26C, rinsed briefly in deionized water, and processed for EM and LM.
Light Microscopy
The cells, grown on coverslips, were dehydrated in ethanol and xylene and then mounted in Canada balsam (BDH; Poole, UK) and were examined and photographed with a Nikon photomicroscope.
Transmission Electron Microscopy
Cells were dehydrated and immersed in a mixture of absolute ethanol and Epon 812 (1:1) and then embedded in pure Epon 812 in the Petri dishes. Beem capsules containing polymerized Epon were placed upside down at a straight angle to the cell layer and were polymerized for 48 hr at 60C. The Epon layer with the Beem capsules was removed from the Petri dish, leaving the cells on the surface of the Epon layer. The Beem capsules were removed with tweezers and ultrathin sections (60 nm) were cut with a diamond knife (DIATOME; Bienne, Switzerland) on a Reichert–Jung ultracut (Vienna, Austria) and collected on Formvar-coated Cu 100-mesh grids. The sections were rapidly counterstained with uranyl acetate and lead citrate and examined in a JEOL 2000-EX TEM electron microscope (Tokyo, Japan) at 100 kV.
Quantitation of Silver Precipitates
Cells were randomly selected at a magnification of X8000 and a video recording of the entire cell was made using a Pixie 8 Image Processor (Deben Research; Suffolk, UK). The recording was transferred to a Macintosh computer and the number and localization of silver-enhanced gold particles were determined. Particles found in vacuolar structures surrounded by a visible membrane structure were classified as lysosomal, and disseminated particles not inside structures with visible membranes were referred to as cytoplasmic (cytosolic). Because the cells varied in size, we calculated the number of silver-enhanced gold particles per 2500 μm2 cell area.
Statistical Evaluation
Specimens of rat heart myocytes originated from different animal preparations. All cultures were immunostained, developed, and embedded at the same time. Silver-enhanced gold particles were quantified in entire cells, and significance was analyzed using the Wilcoxon test.
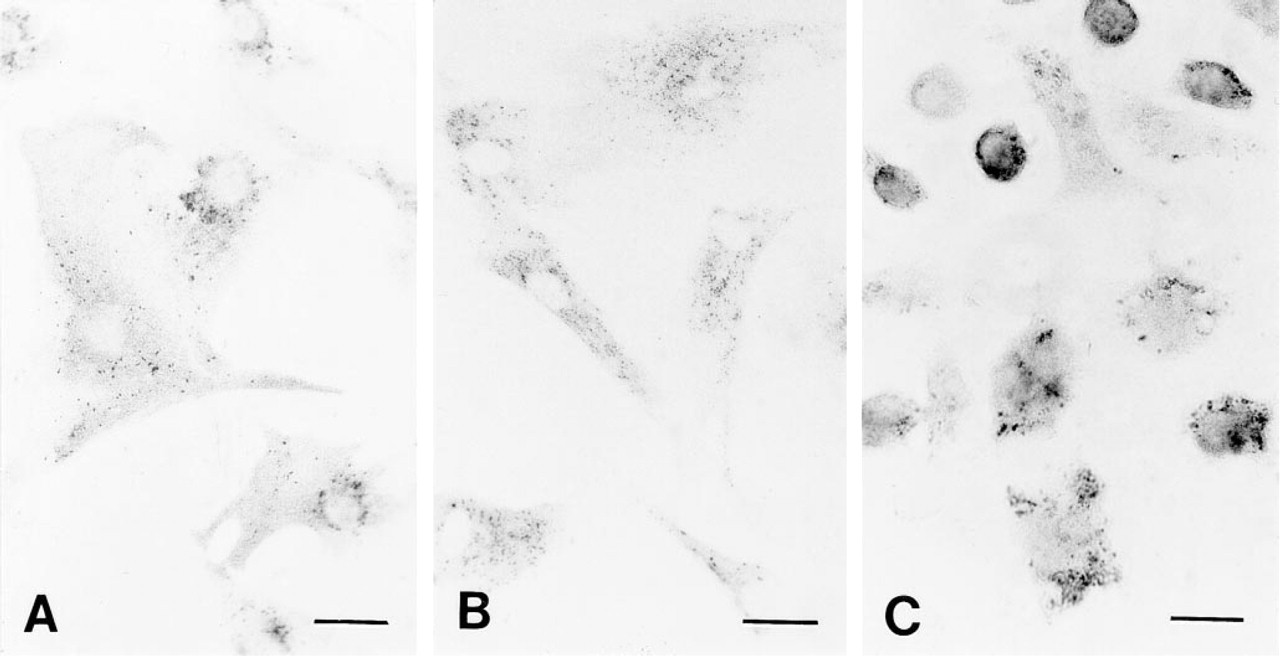
Light microscopy of cells probed with primary polyclonal rabbit anti-cathepsin D antibodies and thereafter with secondary goat anti-rabbit IgG antibodies tagged with 0.8-nm gold particles. The gold particles were enlarged by silver enhancement for 6 min at neutral pH. The micrographs show rat heart myocytes (
Results
As observed in the light microscope, the immunostaining pattern was similar to that noted using immunofluorescence detection of cathepsin D in J-774 cells treated with Triton X-100 during the immuno-incubation (Brunk et al. 1995b) and in fibroblasts permeabilized with 0.1% saponin (Brunk et al. 1997). As shown in Figure 1, fixation followed by sodium borohydride exposure resulted in penetration of the antibodies in all three cell types. The J-774 cells have high phagocytotic activity and they are very rich in cathepsin D, as reflected in a dense precipitate after silver enhancement (Figure 1C). Different combinations of paraformaldehyde (2% or 4%) and glutaraldehyde (1, 0.5, 0.1, or 0.05%) were tested for their effects on ultrastructure and antigenicity. A mixture of 4% paraformaldehyde and 0.05% glutaraldehyde preserved the ultrastructure in such a way that subcellular details were fairly well recognized, i.e., membranes surrounding lysosome-like structures were truly visible. The use of sodium borohydride at 0.05% concentration further improved the penetration of antibodies compared to 0.01%. Moreover, incubation with an F(ab')–gold conjugate instead of IgG–conjugated gold particles did not further increase the labeling density (not shown). Only a few silver precipitates were seen in control cells not exposed to the primary antibody (Table 1; Figure 2A).
Labeling of cathepsin D in rat heart myocytesa

Values represent means of precipitates in a cell area of 2500 μm2 ± SD. The Wilcoxon test showed that values are significantly lower for controls than for the labeled cells, p ≤ 0.01.
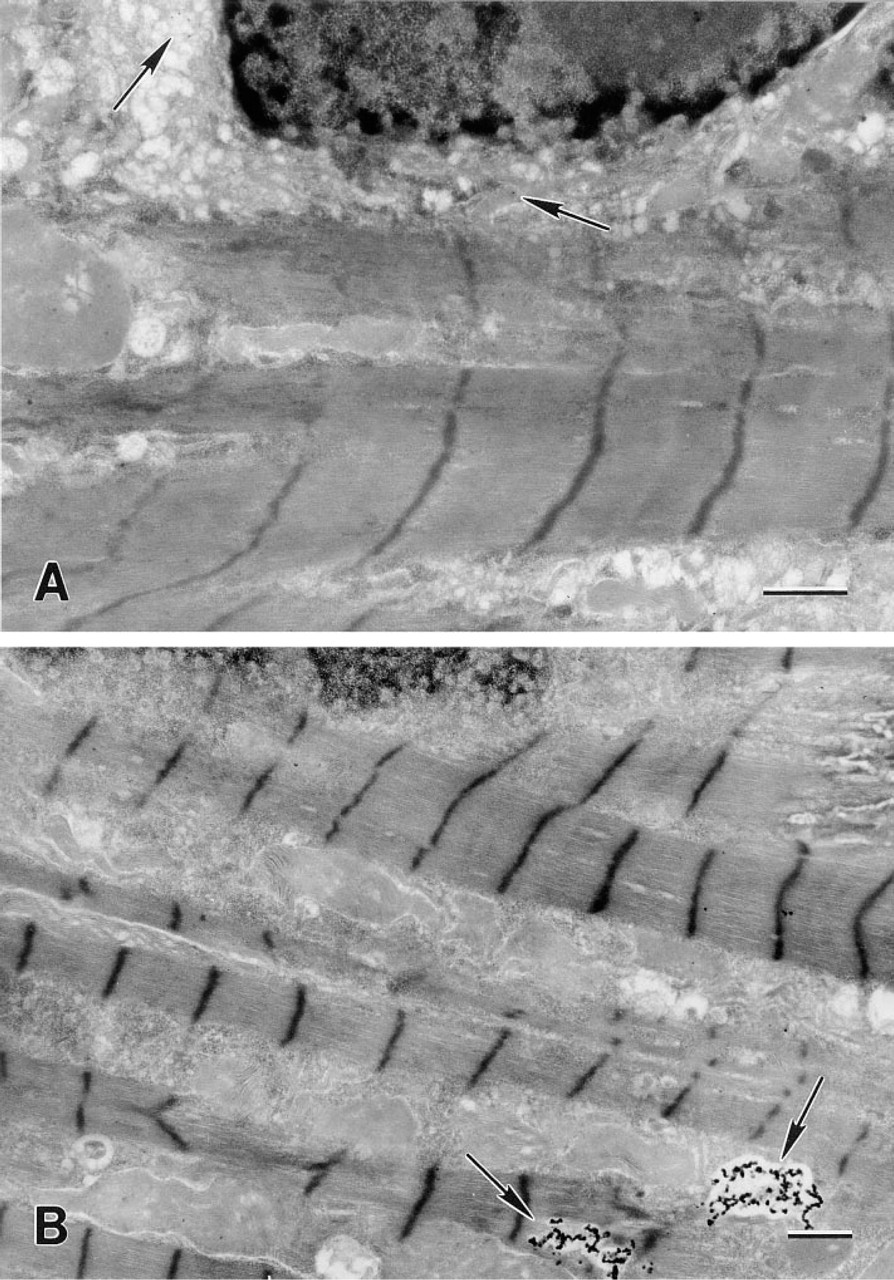
Electron micrograph of rat heart myocytes probed with polyclonal rabbit anti-cathepsin D antibodies and then exposed to goat anti-rabbit IgG antibodies tagged with ultrasmall 0.8-nm gold particles. The gold particles were enlarged by silver enhancement for 6 min at neutral pH. (
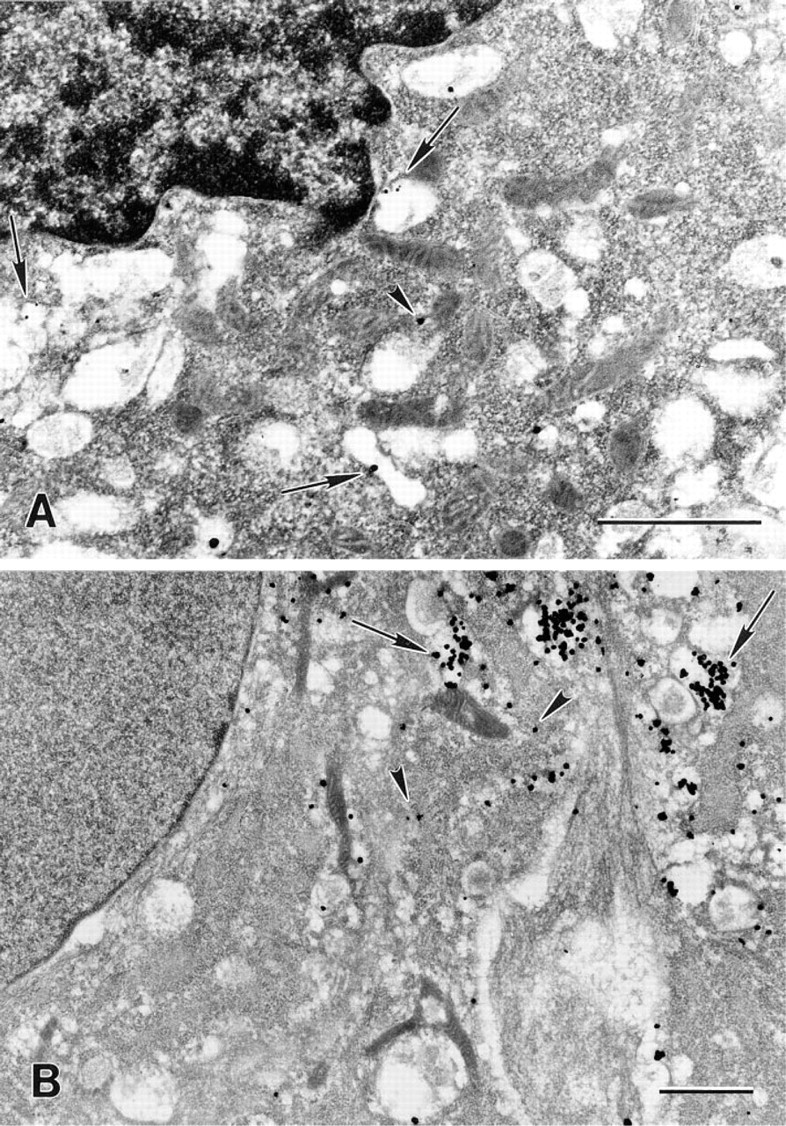
Electron micrographs of mouse histocytic lymophoma (J-774) cells (
Silver development at neutral pH (Lah et al. 1990; Burry et al. 1992) gave reproducible and well-controlled development of the gold particles, with very few unspecific precipitates. Better preservation of membrane structures was also achieved at neutral pH, compared to results obtained at pH 3.5 according to the technique described by Danscher and Norgaard (1983).
Cathepsin D was found primarily in lysosomal structures in the rat heart myocytes (Figure 2B), J-774 cells (Figure 3A), and human foreskin fibroblasts (Figure 3B). Using video images, the silver-enhanced gold particles in the myocytes were counted and registered as being either within lysosomal structures or in the cytosol. As shown in Figure 2A and Table 1, control cells that had been enhanced but had not been exposed to the primary antibody exhibited only a small number of particles, and these were evenly distributed in the cells and showed no statistically significant lysosomal predominance.
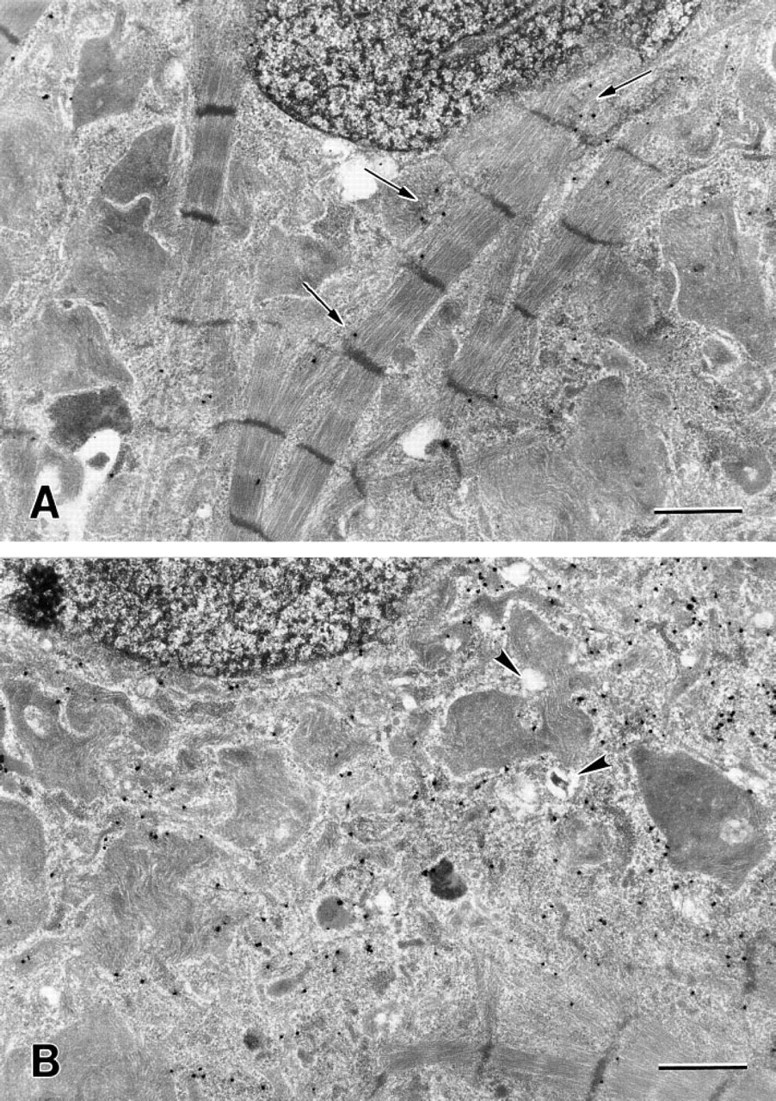
Electron micrographs of rat myocytes after exposure to 5 μM naphthazarin for 30 min (
In all three cell types, oxidative stress caused relocalization of cathepsin D from lysosomal structures to the cytosol. This is illustrated in Figure 4, which shows rat heart myocytes exposed to naphthazarin for 30 and 45 min. Lethal cytotoxicity, measured as decreased plasma membrane integrity (leakage of the enzyme LDH from the cytosol to the surrounding medium), was not detected at this stage but was observed later. Viability was 99, 98.7, and 96.4% for cultures treated with naphthazarin for 0, 30, and 45 min, respectively. After 110 min, 50% of the cells were dead and all cells were devitalized after a couple of hours (not shown).
Discussion
In the present experiments we applied an immunocytochemical pre-embedding immunogold method to different types of cells as a means of localizing the lysosomal aspartic proteinase cathepsin D. Other investigators have used a postembedding immunogold technique to determine the presence of cathepsins B, H, and L in Epon sections of bronchoalveolar epithelial cells (Ishii et al. 1991) and to detect cathepsin D in the lysosomes of human epididymal epithelial cells embedded in Lowicryl K4M (Raczek et al. 1995).
To our knowledge, a pre-embedding method for detection of cathepsin D has not yet been described. Pre-embedding immunogold methods have, however, been used for three-dimensional labeling of nuclear matrix proteins in permeabilized cells (de Graaf et al. 1991,1992) and to study the distribution of the growth-associated protein B-50 in hippocampal neurons (Van Lookeren Campagne et al. 1992). Ultra-small gold particles conjugated to antibodies can penetrate cells that have been subjected to rather mild permeabilization procedures. In one study (Van Lookeren Campagne 1993), the use of gold probes conjugated to the small F(ab')2 was found to improve labeling. Under the conditions used in our experiments, no difference in labeling density was detected in fibroblasts when gold particles conjugated to either F(ab')2 or complete immunoglobulin were used. This was probably due to success of the permeabilization procedure and close to optimal crosslinking.
Amounts of silver-enhanced gold particles were analyzed in heart myocytes that had or had not been exposed to the primary antibody before silver enhancement (Table 1). The negligible background labeling that was observed in the antibody-unexposed control cells was evenly distributed in the cell (Figure 2A), whereas the reaction product was mainly found in the lysosomes of cells subjected to the complete procedure.
Oxygen-derived free radicals damage cellular macromolecules and disrupt cellular homeostasis (Halliwell 1991; Ryan and Aust 1992). Naphthazarin is reduced by intracellular reductases, and the subsequent autoxidation, which utilizes molecular oxygen as an electron acceptor, generates radicals that cause oxidative stress (Cohen and d'Arcy Doherty 1987; Öllinger and Brunmark 1991). However, the role of lysosomes and of lysosomal leakage of potent hydrolytic enzymes in these processes has been largely overlooked.
We have shown in several studies that lysosomal destabilization is an early event in cellular devitalization (Zdolsek et al. 1993; Öllinger and Brunk 1995; Zhang et al. 1995). Nevertheless, even mild and non-lethal exposure of cells to oxidative stress damages lysosomes and causes some enzymatic relocalization, although the damage is repaired (Brunk et al. 1995a). Using immunofluorescence and a confocal laser scanning microscope, we recently showed that cathepsin D is relocalized from an original intragranular (lysosomal) site to the cytosol in J-774 cells (Brunk et al. 1995b), fibroblasts, and myocytes (unpublished observations) exposed to oxidative stress. These results were clearly confirmed by the present findings obtained with the more sensitive pre-embedding immunogold detection technique.
We conclude that our pre-embedding method, in combination with the use of ultrasmall gold probes and ensuing silver enhancement, is a sensitive method for detection of lysosomal enzymes, and offers fairly good morphological preservation that is enough to determine the localization of the antigen. Moreover, this technique should be a very valuable tool for evaluation of lysosomal membrane stability.
Footnotes
Acknowledgements
Supported by the Swedish Cancer Foundation (grant 2703) and by Östgöta läns landsting.
The technical assistance of Ms Britt-Marie Gustafsson is gratefully acknowledged.