
Abstract
TUNEL, i.e., terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling, has become a widely used staining method to assist in detection of apoptotic cells in tissue sections. However, despite its apparent simplicity, this technique has led to considerable disappointment because of its serious limitations in sensitivity and, even more, in specificity. We reviewed the limitations and artifacts of TUNEL and designed a comprehensive protocol to reassess the various procedures in use for five crosslinking and/or precipitating fixatives. By introducing microwave heating in extreme pH-value solutions (pH 3 for formalin and pH 10.6 for Bouin's fixative) coupled with proteolysis, we obtained an intense staining of 70–80% of apoptotic cells and bodies on archival tissue blocks, with little or no background. Owing to the enhanced sensitivity, early stages of apoptosis could be visualized and may enlarge our vision of the apoptotic cell beyond the mere image of shrinkage necrosis. We conclude that TUNEL remains a technique as useful as it is delicate, requiring critical interpretation of the staining. This study points out that, on archival tissues, despite the technical improvements we propose no protocol can be the final answer to all problems. Technique must be readjusted for any variation in tissue processing. However, step-by-step progress has rendered this method not only applicable but also performable within the constraints of archival surgical pathology specimens.
I
By introducing microwaves to TUNEL staining of cultured cells, we obtained considerable gains in sensitivity without impairment of specificity (Negoescu et al. 1996). Taking advantage of an original model, the Basedow-Graves’ goiter, we pursued the same attempt on tissue sections. The study resulted in a tentative explanation of paradoxical stainings and adaptive technical proposals to improve sensitivity and reduce nonspecific labeling, relying on microwave pretreatment at extreme pH values of 3 or 10.6 depending on the fixative.
Materials and Methods
We proceeded in two steps: After having adjusted TUNEL using various fixatives, pretreatments, and labeling protocols on the same Graves’ thyroid specifically prepared for this purpose, the optimized protocol was put to the test of a retrospective investigation of 35 archival cases of Graves’ thyroid. The results, markedly less satisfactory, stressed the extreme sensitivity of TUNEL to tissue processing. Technical investigation was taken up again, now targeted at the two fixatives used for most archival tissues, formalin and Bouin's liquid.
Fixation
Prospective Study. Fragments of thyroid parenchyma measuring 5–8 mm on each side and 3 mm thick (less than 0.5 cm3) were fixed by immersion. Five fixatives were used: 4% (w/v) paraformaldehyde (PFA) in 0.01 M PBS, pH 7.4; 4% (v/v) formaldehyde (formalin) in PBS (pH 7.4); B5 [8% (w/v) mercury chloride in 4% formaldehyde] (pH 6.5); Bouin's liquid [2.5% (w/v) copper acetate, 4% (w/v) picric acid in 3.5% (v/v) formaldehyde, 1.5% (v/v) acetic acid in distilled water] (pH 3.8); and Methacarn [10% (v/v) acetic acid, 30% (v/v) chloroform in methanol] (pH 5.0). Fixation lasted about 20 hr at room temperature (RT) for all fixatives except Methacarn (2 hr).
Retrospective Study. Archival paraffin-embedded specimens measured 1–5 cm2 and were 1.5–3 mm thick. Each had been fixed at RT by immersion, dispatched between formalin and Bouins’ liquid, 20 hr to 4 days, with no specification given as to duration.
Pretreatments
Prospective study. TUNEL was performed with or without pretreatment. For detergent, Triton X-100 (Sigma; Saint Quentin Fallavier, France) was used at 0.1% (v/v) in 0.1% (w/v) sodium citrate (Merck; Darmstadt, Germany) in water for 2 min on ice. Proteinase K (Boehringer; Mannheim, Germany) was used at 20 μg/ml in PBS for 15 min at RT. Other concentrations (2, 10, 20, and 40 μg/ml) and durations (5, 10, and 15 min) were tested.
For microwave irradiation, an AEG Micromat 120 oven (2.45 GHz) with nine power settings (0–850 W) was used. Best results were obtained with Setting 4 (376 W), 5 min in 200 ml of 0.01 M citrate buffer, pH 6 (Lucassen et al. 1995; Sträter et al. 1995), reaching 86C. Tests included variations from Setting 2 (188 W, 54C) to Setting 8 (752 W, 99.6C; boiling) for one or two 5-min periods; we also compared classical 15-min progressive cooling in the citrate buffer (Shi et al. 1995) versus rapid cooling (Negoescu et al. 1996).
As a combination pretreatment (Sträter et al. 1995); the slides were irradiated in 50 ml of citrate buffer for 1 min at 750 W (86C), rapidly cooled, and then incubated for 15 min at RT in 20 μg/ml proteinase K.
No inhibition of endogenous peroxidase was performed because H2O2 weakens TdT activity (Migheli et al. 1995) and induces DNA breaks (Wijsman et al. 1993).
Retrospective Study. Detergent treatment was performed as in the previous step but was always combined with other pretreatments. The proteolytic enzyme was used at 15, 20, 25, or 40 μg/ml for 15 min at RT. For microwave irradiation, the slides (8 to 15) were immersed in 200 ml of the following solutions: 0.01 M citrate buffer (pH 3 and 6), 0.01 M sodium hydrogen carbonate (Merck; pH 9), 0.05 M tris-[hydroxymethyl]aminomethane (Tris; Sigma, pH 10.6) or a 1/1 (v/v) mixture of citrate buffer (pH 6) and Tris solution (pH 10.6) to a final pH of 9. Citrate buffer and the citrate/Tris mixture were heated for one or two cycles of 5 min at Setting 8 (752 W). To avoid section detachement in carbonate and Tris solutions, slides were first irradiated for 3 min at Setting 8 and then maintained at Setting 4 for 2–22 min. Boiling temperature was 99.6C for all solutions, reached in 3 min at Setting 8.
Pretreatment combinations included the following: (a) Triton followed by proteinase K (15, 20, and 25 μg/ml); (b) microwaving followed by Triton; (c) microwaving followed by proteinase K (15, 20, and 25 μg/ml); and (d) microwaving followed by Triton and then by proteinase K (15, 20, and 25 μg/ml).
TUNEL
Prospective Study. Three modalities of TUNEL were tested: a laboratory protocol and kits from Boehringer Mannheim (In Situ Cell Death Detection Kit, Peroxidase) and from Oncor (ApopTag Plus; Gaithersburg, MD). The laboratory protocol was adapted from Gavrieli et al. (1992), Gold et al. (1994), and Migheli et al. (1994) and was performed with Boehringer Mannheim reagents. After a 30-min (RT) incubation with 3% BSA, 20% normal bovine serum (Jackson; West Grove, PA) in PBS, pH 7.4, sections were covered with the TUNEL mix [0.135 U/ml calf thymus TdT, 0.0044 nmol/ml digoxigenin-11-2'dUTP, and 1 mM cobalt chloride in 1 X reaction buffer (200 mM potassium chloride, 25 mM Tris-HCl, 0.25 mg/ml BSA in distilled water, pH 6.6)] for 1 hr at 37C. After another saturation [30 min at RT in 3% BSA and 20% normal sheep serum (Jackson) in 1% (w/v) blocking reagent (Boehringer Mannheim) in 0.1 M Tris-buffered saline], the sections were treated with a peroxidase-labeled digoxigenin sheep Fab antibody (1.25 peroxidase U/ml) for 1 hr at RT. The Boehringer Mannheim kit was used according to the manufacturer's instructions with the addition of the two normal-serum saturations mentioned above, since this kit uses calf thymus TdT. A 1:3 dilution of the antibody yielded an optimal signal-to-background ratio. The Apop-Tag kit was used without saturations because neither the origin of the TdT nor that of the antibody was provided. Reaction with 0.05% 3–3'-diaminobenzidine tetrahydrochloride (DAB) was monitored under the microscope and lasted from 10 sec to 2 min. Negative control was performed by omitting TdT. As a positive control we used paraffin-embedded sections of 10% buffered formalin-fixed weaning mammary glands (ApopTag Control Slides; Oncor). After counterstaining with Harris’ hematoxylin, slides were dehydrated and mounted in Merckoglas (Merck).
Retrospective Study. The Boehringer Mannheim kit alone was used.
Cell Counting and Statistical Analysis
Prospective Study. Once established, each optimal method was reproduced in triplicate and quantification was performed on a slide representative of each working condition. Thyreocytes in the vesicle lumina were assessed for morphology and staining, according to Gold et al. (1994). The following findings were considered to represent apoptosis (Kerr et al. 1972; Wyllie et al. 1980): (a) marked condensation of chromatin and cytoplasm (apoptotic cells); (b) cytoplasmic fragments with or without condensed chromatin (apoptotic bodies); and (c) intra- and extracellular chromatin fragments (micronuclei). Values represent percentages from at least 1000 counted apoptotic and non-apoptotic cells. Using the Fisher's exact test (Haycock et al. 1992), differences were considered as significant at p<0.05.
Retrospective Study. The tissue sections were about 10 times larger than those used in the first step and we noted uneven labeling from one area to another. Therefore, a semiquantitative evaluation was performed, with four parameters (Table 1): fraction of apoptotic cells labeled; labeling intensity; non-apoptotic cell coloration; and connective tissue background.
Results
Prospective Study (see Figure 1)
TUNEL Protocols. The three modalities of TUNEL we tested (laboratory protocol, ApopTag kit, and Boehringer Mannheim kit) yielded no significantly different results, with the exception of proteinase K-pretreated tissue sections fixed with either Bouin or Methacarn, for which the Boehringer Mannheim kit proved more sensitive than the two others, having a good signal-to-background ratio and being the easiest to use; therefore, it was used for the retrospective study.
Fixatives, Cells, and Labeling Morphology. The highest resolution of nuclear morphology was provided by Bouin's fluid (Figure 2D). Nevertheless, no difficulty was encountered in identifying the criteria of Kerr and Wyllie for apoptotic cells, no matter what the fixative. Some of the apoptotic bodies in the vesicle lumina were in secondary necrosis with loss of nuclear and cytoplasmic outlines. These were not included in the counts.
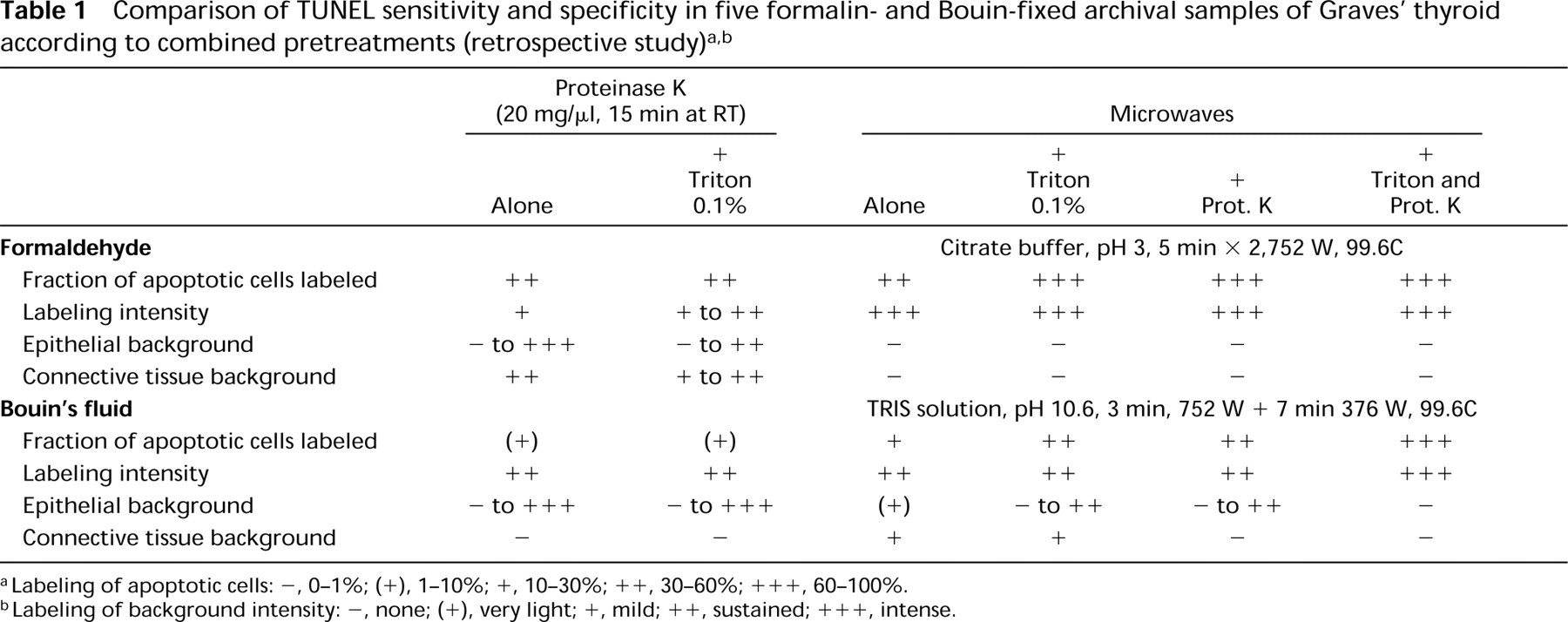
Labeling of apoptotic cells: -, 0–1%; (+), 1–10%; +, 10–30%; ++, 30–60%; +++, 60–100%.
Labeling of background intensity: -, none; (+), very light; +, mild; ++, sustained; +++, intense.
Effect of Pretreatments. TUNEL performed without any pretreatment yielded unsatisfactory results, with 8–51% of apoptotic-appearing cells being stained. The best pretreatment proved to be proteinase K (20 μg/ml, 15 min at RT) (p<0.05), with about 70% labeled apoptotic cells for aldehyde fixatives and Methacarn and about 60% for Bouin's fluid, without increasing the proportion of morphologically normal stained cells. Triton and microwaving led to a smaller gain in specific staining (p<0.05) while increasing the percentage of labeled cells with a non-apoptotic appearance. Combined microwave–proteinase K pretreatment (according to Sträter's protocol) did not prove to be more efficient than proteinase K alone (p<0.05). No important differences were noted between classically and rapidly cooled slides. Therefore, classic cooling (15 min) was preferred for its convenience.
TUNEL and fixatives (prospective study). (
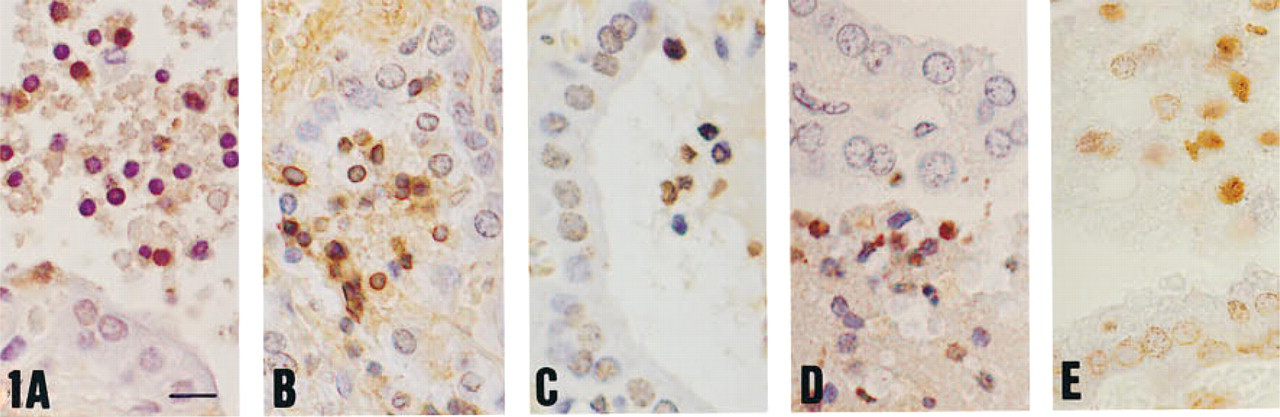
TUNEL drawbacks and improvement in archival Graves’ thyroid specimens (retrospective study; see also Figure 3). (
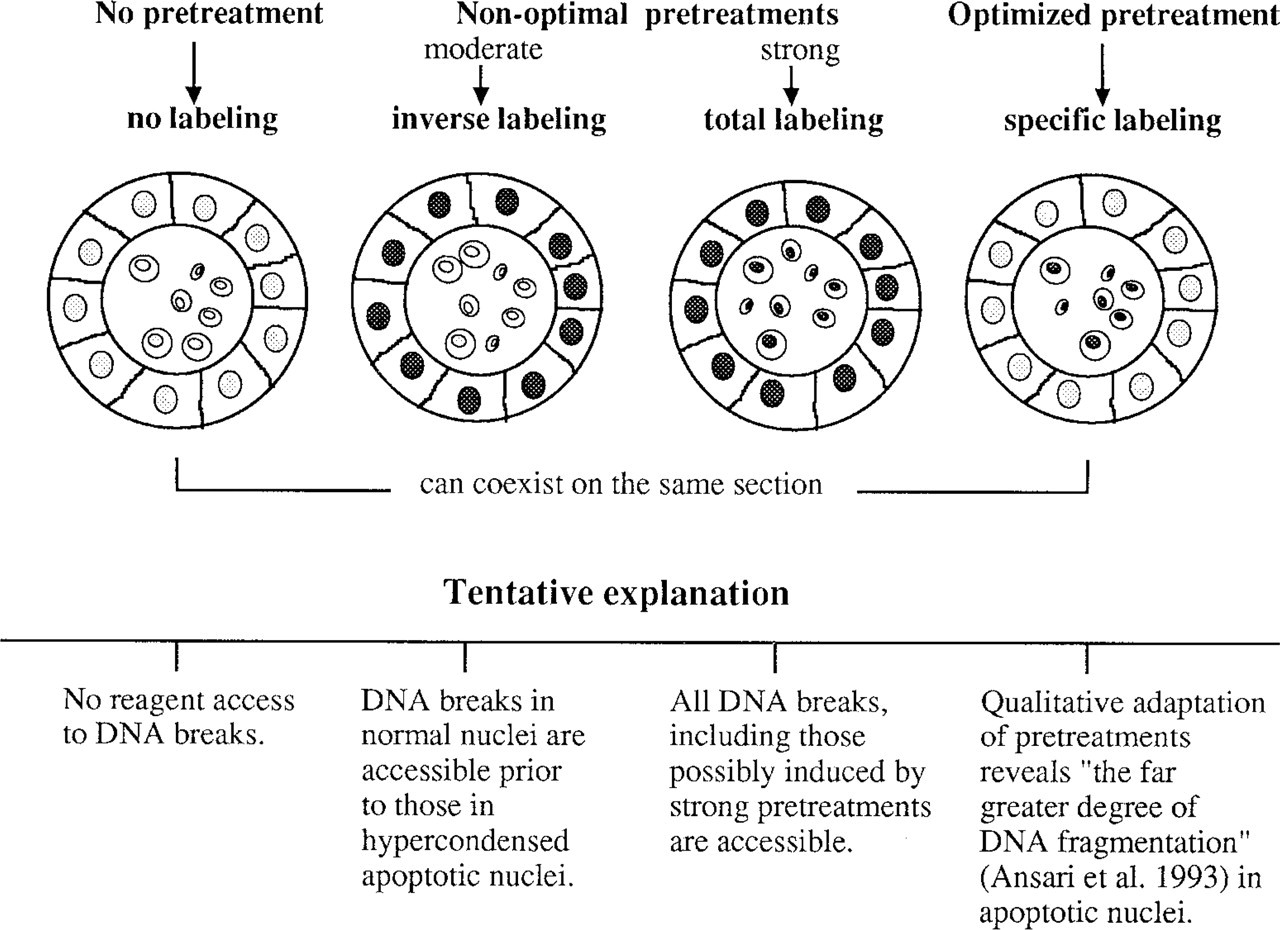
Scheme of TUNEL patterns and pitfalls in relation to tissue treatment. Apoptosis is characterized by strong DNA compaction and a high degree of DNA fragmentation. Tissue fixation, i.e., protein and DNA crosslinking and denaturation, hinders TUNEL reagent access to the 3'OH recessed ends. In addition, normal DNA is also exposed to natural and artifactual breakage. Empirically optimized pretreatments were aimed at giving access of reagents to DNA breaks. Monitoring of the color reaction completes this by identifying the moment at which staining emerges at the numerous labeled sites in apoptotic cells, before the appearance of the much less labeled normal DNA.
Controls and Background Treatment (data not shown). Lack of TdT in the TUNEL mix abolished labeling. Positive controls exhibited many stained, condensed nuclei and micronuclei, either isolated or within the cytoplasm of phagocytic cells. The connective stroma of the thyroid was colored by TUNEL, sometimes intensely. This collagen-bound background was abolished in TdT-lacking controls. Therefore, to investigate the possibility of calf TdT-collagen interaction, we introduced a 20% normal bovine serum incubation before TUNEL, which significantly decreased background.
Retrospective Study (Table 1; Figures 2 and 3)
The protocol retained as best adapted (proteinase K 20 μg/ml, 15 min at RT, followed by TUNEL performed with the Boehringer Mannheim kit) to light fixation of small tissue blocks, when applied to an archival series of 35 formalin- and Bouin-fixed Graves’ thyroids, yielded disappointing results. Overall, three situations were encountered, either alone or combined on the same section: no staining, general staining of all nuclei (apoptotic or not), or “inverse staining,” i.e., lack of or low staining of apoptotic cells surrounded by intense staining of virtually all normal nuclei (Figures 2A and 2B). At best, the sensitivity was about 40% in formalin-fixed tissues and was less than 10% in Bouin-fixed samples (Figurea 2C and 2D). TUNEL was not improved by varying proteinase K concentration or by adding a detergent step to the enzymatic digestion. For microwaving, of the various pH values investigated with different irradiation times, the following did not improve TUNEL when used either alone or combined with Triton, proteinase K, or both: the carbonate solution (pH 9) altered the histological structures and frequently detached sections; the citrate/Tris mixture (same pH of 9 but less aggressive for the tissues); and citrate buffer (pH 6; Figures 2E and 2F).
Significant improvement was achieved when extreme pH values were explored (Figures 2G and 2H). For formalin-fixed samples, citrate buffer (pH 3), twice for 5 min at 752 W, abolished background. Sensitivity remained stable compared to proteinase K digestion, and therefore the readability of the labeling was improved.
An increment in sensitivity to about 80% of apoptotic cells was achieved by addition of a second pretreatment with proteinase K (20 μg/ml). Labeling morphology was influenced by pretreatments. In the absence of proteolysis, nuclear labeling was faint and the cytoplasm of apoptotic cells was sometimes darker than the nuclei. Triton favored staining of apoptotic cell cytoplasm, together with an extremely clean general background. Proteinase K enabled the labeling to concentrate in the nuclei, the cytoplasm being either stained or unstained. Therefore, we adopted acidic pH and the three-pretreatment combination as our best protocol for formalin-fixed tissues. For Bouin-fixed sections, a similar improvement resulted from the use of the basic Tris solution (pH 10.6) with a comparable energy input (3 min at 752 W plus 7 min at 376 W), followed by Triton and proteinase K (20 μg/ml). The analysis of apoptosis in the 35-case series treated according to these protocols will be reported in the future (Labat-Moleur et al., manuscript in preparation). The DAB color reaction was optimal when it was short (10 sec to 1 min) and was carefully monitored, under the microscope, slide by slide, to avoid background.
Thyreocytes with large nuclei (about 1.5 times as large as those of surrounding cells) and with a thin rim of marginated and condensed chromatin were observed in the vesicular epithelium in the vicinity of highly apoptotic foci. These dilated nuclei were quite numerous and their chromatin rim was as intensely TUNEL-positive as that of condensed apoptotic bodies. These cells may represent an early stage of apoptosis (Figure 2I).
In summary, among the fixation variations of our archival blocks, the best results were obtained with combined pretreatment: (a) microwave irradiation (around 10 min) at acidic pH (pH 3) for aldehyde-fixed tissues and basic pH (pH 10.6) for Bouin's-fixed tissues; and (b) proteinase K, 15 min at 20 μg/ml, the conditions used by most authors. Indeed, our tests revealed no gain when this parameter was varied. Triton proved to be optional. The optimal color reaction was brief, 10 sec to 1 min.
Discussion
We attempted in this investigation to overcome the main drawbacks of TUNEL detection of apoptotic cells in tissue sections. Several authors (Ansari et al. 1993; Grasl–Kraupp et al. 1995; Mundle and Raza 1995; Sasano 1995; Yasuda et al. 1995; Cervos–Navarro and Schubert 1996) have expressed limited confidence in TUNEL because of severe shortcomings in sensitivity and even more in specificity because TUNEL is blamed for staining, in addition to necrosis (Grasl–Kraupp et al. 1995), of “a large number of the cells in which apoptosis can hardly occur, including proliferative cells” (Sasano 1995).
We suggest separating artifactual staining of necrosis and mitosis from the body of criticism. Several authors (Gorczyca et al. 1993; Migheli et al. 1995; Brambilla et al. 1996) have observed that only a minority of necrotic and mitotic cells were TUNEL-positive in tissues, as opposed to the frequent staining by TUNEL of cultured mitotic cells (Negoescu et al. 1996). Moreover, if labeled, these can quite easily be distinguished from apoptotic cells on morphological grounds, particularly because DNA is broken at late stages of necrosis (Gold et al. 1994) when morphological alterations are unambiguous. As for mitoses, although chromatin condensation at telophase may mimic apoptosis (Migheli et al. 1994,1995), the greatest analogy between mitotic and apoptotic aspects occurs in abortive mitoses, a derivation of cell division towards active cell death sometimes named “mitotic catastrophe” (Martin et al. 1995).
The major problem with TUNEL, as recurrently emerging in the critical evaluations devoted to the technique and confirmed by our results, can be summarized as follows (Figures 2 and 3). (a) Without pretreatments, TUNEL sensitivity is far too low. (b) Pretreatments with acknowledged efficiency, i.e., proteinase K and microwaves, easily induce general labeling of morphologically normal nuclei (Migheli et al. 1994; Lucassen et al. 1995). (c) TUNEL proved to be very sensitive to fixation (Lucassen et al. 1995), which makes sample size critical. Although fixation is homogeneous in small tissue blocks (5 mm2, as in our prospective study), fixation is uneven from the periphery to the central area of large samples, which are the rule in archives. (d) Presumably, when combined, these three factors may contribute to the presence in the same tissue section of blank areas, together with specifically labeled spots and areas in which all nuclei are stained.
This points up the fact that TUNEL is an ambitious approach whose target, apoptotic DNA breaks, is less accessible than breaks that occur in non-apoptotic DNA. In fact, many known causes of DNA breaks can be listed: DNA recombination, replication, repair or compaction–relaxation during mitosis, tissue electrocoagulation, autolysis, fixation, paraffin embedding, cutting, and pretreatments with H2O2, detergent, proteinase K, and microwaves (Szabò et al. 1990; Eastman and Barry 1992; Ansari et al. 1993; Wijsman et al. 1993; Migheli et al. 1994; Lucassen et al. 1995; Sasano et al. 1995; Wolffe 1995; Negoescu et al. 1996). Strong DNA compaction, a hallmark of the mechanism of apoptosis, combines with protein and DNA crosslinking and with precipitation induced by fixation to hinder reagent access to the 3'OH recessed ends. However, TUNEL has an advantage: the far greater degree of apoptotic DNA fragmentation (Ansari et al. 1993). How can we obtain a good differential staining between apoptotic and non-apoptotic DNA, given that all means of break disclosure (detergents, proteases, microwaves) are themselves able to create breaks (Szabò et al. 1990; Migheli et al. 1994; Negoescu et al. 1996)? On the basis of our extensive tests, it appears that the answer does not come from retrieval reinforcement but from qualitative adaptations of retrieval techniques, as illustrated by the opposite pHs that respectively rescued TUNEL specificity in formalin- and Bouin-fixed tissues (Table 1). Shi and co-workers have already noted the impact of pH on microwave retrieval efficiency (Shi et al. 1995). Interestingly, they observed that nuclear antigens are best retrieved at extreme pH (around 2 and 10) while admitting that the mechanism of this phenomenon is not clear.
Even when optimal, pretreatments do not guarantee lack of background. Therefore, we agree with the trend to carefully control the detection system (Migheli et al. 1994): dilution of the enzyme-coupled antibody, choice of enzyme, and monitoring of the color reaction. Lack of color reaction standardization implies limiting quantification to those cells whose morphology supports apoptosis. At any rate, labeling intensity must not be taken into account: every nucleus stained, weakly or strongly, is to be scored, provided that it is concordant with morphological criteria.
When fixation is controlled and can be kept homogeneous and light, as in prospective studies, proteinase K alone may be sufficient for all the crosslinking aldehyde fixatives we tested (paraformaldehyde, formalin, B5), with more than 70% apoptotic cells labeled (Figure 1). When tissues were fixed with precipitating solutions (Methacarn, Bouin) only about half of the apoptotic cells were stained. A comparison of proteinase K efficiency between the prospective and retrospective steps illustrates the dramatic drop in sensitivity (75% to 40%, 57% to 5% for Bouin) linked to the variation of block size and fixation duration (frequently drastic overfixation). In this situation, the combined use of microwaves plus proteinase pretreatment suggested by Sträter et al. (1995) proved adequate, provided that extreme pH values are introduced.
As a consequence of enhanced sensitivity and specificity of TUNEL staining, the constant and intense labeling of dilated nuclei within a peculiar environment of vesicular remodeling adjacent to apoptosis-rich areas (manuscript in preparation) can be accepted as specific (Figure 2I). This image probably corresponds to the “swelling cells,” stage 3 of the model proposed by Desjardins and MacManus (1995), and to the aspects mentioned by Nishikawa and Sasaki (1995) and Skoff (1995). At this step, both chromatin condensation and fragmentation are at their beginning, producing large DNA fragments (around 400 base pairs) (Arends et al. 1990; Cohen et al. 1993). By tracking the apoptotic sequence up to its early stages, TUNEL may be enlarging our vision of the apoptotic cell beyond the picture of shrinkage necrosis.
In conclusion, despite its hardly accessible target, step-by-step TUNEL improvements enable accurate identification of apoptosis, even within the constraints of archival surgical pathology specimens. Nevertheless, TUNEL is so sensitive to tissue processing that no unique protocol can be proposed. Whenever possible, fixation must be standardized. For archival samples, adaptative tests must be performed for each particular case series. Standardization of TUNEL is a critical issue that requires further studies based on control tissues or cell lines with known quantitative information concerning apoptosis. Such control tissues should be used to monitor the TUNEL procedure so as to achieve more accurate delineation of apoptosis.
Footnotes
Acknowledgements
A. Negoescu acknowledges financial support from the COMARES (Comité des Maladies Respiratoires de l'Isère).
We thank Dr Ruth Griffin–Shea, who kindly revised the English version of this manuscript. We are indebted to Catherine Pépin and Jean-Michel Lasserre for the iconography.