
Abstract
Methylmethacrylate (MMA) embedding of undecalcified bone biopsies is a technique widely used for bone histomorphometry. However, conventional MMA embedding causes almost complete loss of enzyme activity and protein antigenicity in the tissues. Recently, an MMA embedding technique has been reported that preserves enzyme activity and antigenic determinants in bone tissue. We describe here a modification of this embedding method. For our modified MMA embedding process, commercially available methacrylates can be used without purification, and the histologic quality of bone sections is comparable to that of conventionally MMA-embedded bone specimens. The technique reported here can be employed for embedding of larger bone samples and is suitable for histochemical and immunohistological applications as well as for routine bone histomorphometry. By addition of methylbenzoate during infiltration and polymerization of the plastic, the antigenicity of the tissue was improved. As applications of this novel technique, demonstration of alkaline phosphatase and tartrate-resistant acid phosphatase as well as positive labeling of Kupffer cells and osteoclasts with the monoclonal antibody ED1 in sections of liver, tibiae, and vertebrae of 3-month-old rats was demonstrated. The method described here might be useful for the inclusion of histochemical and immunohistological methods into bone histomorphometry.
Keywords
M
Polymerization of methacrylates is an exothermic radical chain reaction. Conventional MMA mixtures mostly use organic peroxides as initiators of polymerization, and polymerization is usually carried out at temperatures between 30 and 45C, using thermal decomposition of the peroxide to start the polymerization process (Schenk et al. 1984; Baron et al. 1983). It is believed that enzyme activity and antigenic determinants are lost because of temperature peaks and modification of chemical structures in the tissue by reactive radicals formed during polymerization of MMA (Baskin et al. 1992; Wolf et al. 1992). It is also possible to initiate polymerization of methacrylates by blue or
In vivo labeling of bone with fluorochromes (e.g., tetracyclines) is a widely used method to assess mineral apposition rate and bone formation rate in bone histomorphometry. Fluorochromes are calcium-seeking molecules that bind to the mineralization fronts in bone formation sites. In sections of undecalcified bone, fluorochromes can be visualized by their specific fluorescence under
Recently, an MMA embedding technique has been reported that can be used for embedding of larger bone samples, and which preserves enzyme activities and antigenic determinants in bone tissue (Wolf et al. 1992). This embedding mixture polymerizes at −15 to −20C. It employs benzoyl peroxide as initiator and the aromatic amine N,N-dimethyl-p-toluidine as accelerator for chemical polymerization at low temperatures. However, the described MMA embedding technique involves tedious purification and destabilization of the methacrylate components, and the histological quality of the sections, in our hands, is inferior in comparison to specimens embedded with conventional MMA techniques.
We report here a modification of the embedding technique originally described by Wolf et al. (1992). For this modified embedding process, commercially available methacrylates can be used without purification, and the histological quality of bone sections is comparable to that of conventionally MMA-embedded bone specimens. As examples of two histochemical reactions relevant to bone research, positive staining of osteoblasts for alkaline phosphatase (AP) and of osteoclasts for tartrate-resistant acid phosphatase (TRAP) in bone sections is shown. As example of an immunohistochemical reaction, positive labeling of rat macrophages and osteoclasts with the monoclonal antibody (MAb) ED1 is demonstrated in sections of rat liver and bone.
Materials and Methods
Animal Procedures
Six 3-month-old female Fischer-344 rats (Charles River; Sulzfeld, Germany) were used for this study. The rats were housed in pairs at 24C and a 12-hr/12-hr light/dark cycle with free access to tapwater and a commercial rat diet (Altromin 1320; Altromin, Lage, Germany). On Days 14, 9, and 4 before sacrifice, each rat was injected
Histology
If not otherwise specified, all chemicals were purchased from Merck (Darmstadt, Germany). All steps of fixation, dehydration, and infiltration were carried out at 4C on a magnetic stirrer. The tissue samples were fixed in 40% ethanol for 48 hrs or in 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer, at pH 7.4, for 24 hr. The tissue specimens fixed in PFA were washed overnight in 0.1 M phosphate buffer, pH 7.4, containing 10% sucrose. Subsequently, the bone and liver samples were dehydrated according to the following schedule (Schenk et al. 1984): 70% ethanol (2 days), 95% ethanol (2 days), 100% 2-propanol (twice for 1 day), and xylene (twice for 2 days). After dehydration, the samples were infiltrated with the plastic embedding mixture using a three-step protocol. In each MMA solution, the samples were infiltrated for 3 days. MMA Solution I consisted of 60 ml MMA (Merck; containing 100 ppm hydrochinon) + 35 ml butylmethacrylate (Sigma; containing 10 ppm hydrochinon) + 5 ml methylbenzoate + 1.2 ml polyethylene glycol 400. MMA Solution II was a mixture of 100 ml MMA I with 0.4 g dry benzoyl peroxide, and MMA Solution III consisted of 100 ml MMA I + 0.8 g dry benzoyl peroxide. All MMA solutions were stirred for at least 1 hr before use. Polymerization was carried out in 25-ml glass vials (if the sample size permits, smaller glass vials can be used without modification of the method). The infiltrated tissue was placed on a polymerized layer of plastic in the bottom of the glass vials. To prepare the plastic bases, 600 μl of N,N-dimethyl-p-toluidine was added to 100 ml of cold (4C) MMA III and stirred for a few minutes. Immediately thereafter, 5 ml of the polymerization mixture was poured into each glass vial. Each vial was then thoroughly gased with N2 or CO2, capped, and polymerized within 1 day at 4C. The polymerization process was sensitive to oxygen, and bases did not polymerize in the presence of oxygen. The glass vials with the plastic bases can be stored for years. To prepare the polymerization mixture, 400 μl of N,N-dimethyl-p-toluidine was added to 100 ml of cold (4C) MMA III, and stirred for a few minutes. After addition of the accelerator, care was taken that the polymerization mixture was kept cold at all times. After the infiltrated tissue was placed on the plastic layer in the vials, the vials were completely filled with polymerization mixture (about 20 ml) to exclude air, capped, and transferred to a deep-freezer. Polymerization was carried out at −18 to −20C and was complete within 3 days. Polymerized blocks were stored at −20C.
After trimming of the plastic blocks, 3-5-μm thick sections were prepared at room temperature with a Microm HM 360 microtome (Microm; Walldorf, Germany) equipped with a D profile knife with a tungsten carbide cutting edge. During sectioning, the knife and the blocks were kept moist with a sectioning fluid (WIV; Schwetzingen, Germany) or with 30-40% ethanol. The sections were transferred onto chromalum-gelatin-coated slides (for conventional staining and fluorochromes) or to slides pre-treated with 3-amino-propyltriethoxy-silane (APES, Sigma; for histochemistry and immunohistochemistry) and carefully stretched using 70% ethanol. Thereafter, the sections were covered with a polyethylene foil, flattened with a rubber roller, pressed with a slide press, and dried for 2 days at 42C in the slide press.
For deplasticization, the sections were placed in three changes of 2-methoxyethylacetate for 20 min each, two changes of acetone for 5 min each, and two changes of deionized water for 5 min each. Sections were routinely stained with toluidine blue at acid pH (Baron et al. 1983) and with von Kossa (Schenk et al. 1984). Sections for analysis of fluorochrome labeling were not deplasticized, were left unstained, and were mounted with Fluoromount (Serva; Heidelberg, Germany).
Histochemistry
For demonstration of AP activity, deplasticized sections were transferred into 0.1 M Tris-HCl buffer, pH 8.2, for 5 min, and the histochemical reaction was then carried out in 0.1 M Tris-HCl buffer, pH 8.2, using a commercially available kit (Vector Red; Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions. Positive staining developed within 30 min to 2 hr at 37C. Control sections were incubated in incubation medium without the enzyme substrate. No staining developed in these control sections.
For TRAP histochemistry, deplasticized sections were placed in 0.1 M acetate buffer, at pH 5.0, for 5 min, and the TRAP reaction was subsequently performed as described previously by Baron et al. (1983) using hexazotized pararosaniline (Merck) as azo dye and naphthol AS-TR phosphate (Sigma) as enzyme substrate. Tartaric acid was added to the incubation medium at a concentration of 100 mM. Positive staining developed within 15 min to 2 hr at 37C. Control sections were incubated in incubation solution that did not contain the enzyme substrate. No staining developed in these control sections.
Counterstaining for sections stained for AP and TRAP was performed using Mayer's hematoxylin (1 min), and the sections were subsequently mounted with an aqueous mounting medium (Kaiser's glycerol gelatin; Merck).
Immunohistochemistry
Immunohistochemistry was performed using the alkaline phosphatase-anti-alkaline phosphatase (APAAP) method. Deplasticized sections were placed in 0.1 M Tris-HCl buffer, at pH 8.2, for 5 min. After blocking the sections with 10% rabbit serum (Vector) for 20 min, the sections were incubated overnight at 4C with the MAb ED1 (Serotec; Oxford, UK) at a 1:100 dilution in Tris-buffered saline (TBS; 0.1 M Tris, 0.15 M NaCl, pH 8.2) with 1% rabbit serum. Control sections were incubated with a non-immune myeloma IgG1 (clone MOPC-21; Sigma) at the same concentration as the ED1 antibody. After washing in TBS containing 0.1% Tween 20 (three times for 5 min), the sections were incubated for 30 min with rabbit anti-mouse antibody (STAR 37, not crossreacting with rat; Serotec) at 1:25 dilution in TBS with 1% rabbit serum. After washing (three times for 5 min in TBS with 0.1% Tween 20), sections were incubated for 30 min with mouse monoclonal APAAP complex (Dako; Hamburg, Germany) at 1:50 dilution in TBS. Thereafter, the sections were rinsed (three times for 5 min in TBS with 0.1% Tween 20), and stained with the Vector Red kit according to the supplier's instructions. Levamisole (Vector) was added to the incubation medium at the concentration recommended by the supplier. Finally, the sections were counterstained with Mayer's hematoxylin for 1 min and mounted with an aqueous mounting medium (Kaiser's glycerol gelatin).
Results
The embedding protocol employed in this study resulted in reliable and homogeneous polymerization of the blocks. The tendency to form bubbles during polymerization at the interface between the glass vial and the tissue surface was reduced when glass vials with a pre-polymerized base were used. Hardness of the blocks, sectioning properties, stretching of the sections, and section quality were comparable to those of conventionally MMA-embedded bone specimens. The hardness of the blocks did not increase even after prolonged storage at room temperature (months to years). The staining characteristics of the sections were excellent. The staining protocols used for sections of conventionally MMA-embedded bones could be used without any modification. Compared to fixation with 40% ethanol, PFA fixation resulted in better morphological detail, with little shrinkage of bone marrow cells (Figure 1). In vivo fluorochrome labeling of the bones could be visualized by fluorescence microscopy. The intensity of the fluorescence signals was independent of the fixation used and was comparable to that of specimens embedded in conventional MMA (Figure 2).
Osteoclasts on all periosteal and endosteal bone surfaces stained positive for TRAP activity (Figure 3). TRAP-positive cells not in direct contact or in close proximity to bone surfaces occurred only very rarely in bone marrow. The intensity of TRAP activity was about the same in 40% ethanol- and PFA-fixed specimens. AP activity, on the other hand, was more sensitive to different fixation processes and was greatly reduced by PFA fixation. Osteoblasts as well as many leukocytes in bone marrow stained positive for AP in specimens fixed with 40% ethanol at 4C (Figure 4). Cells positive for AP showed a membranous staining pattern in most cases. For both TRAP and AP, staining was absent in control sections incubated in medium lacking the enzyme substrate.
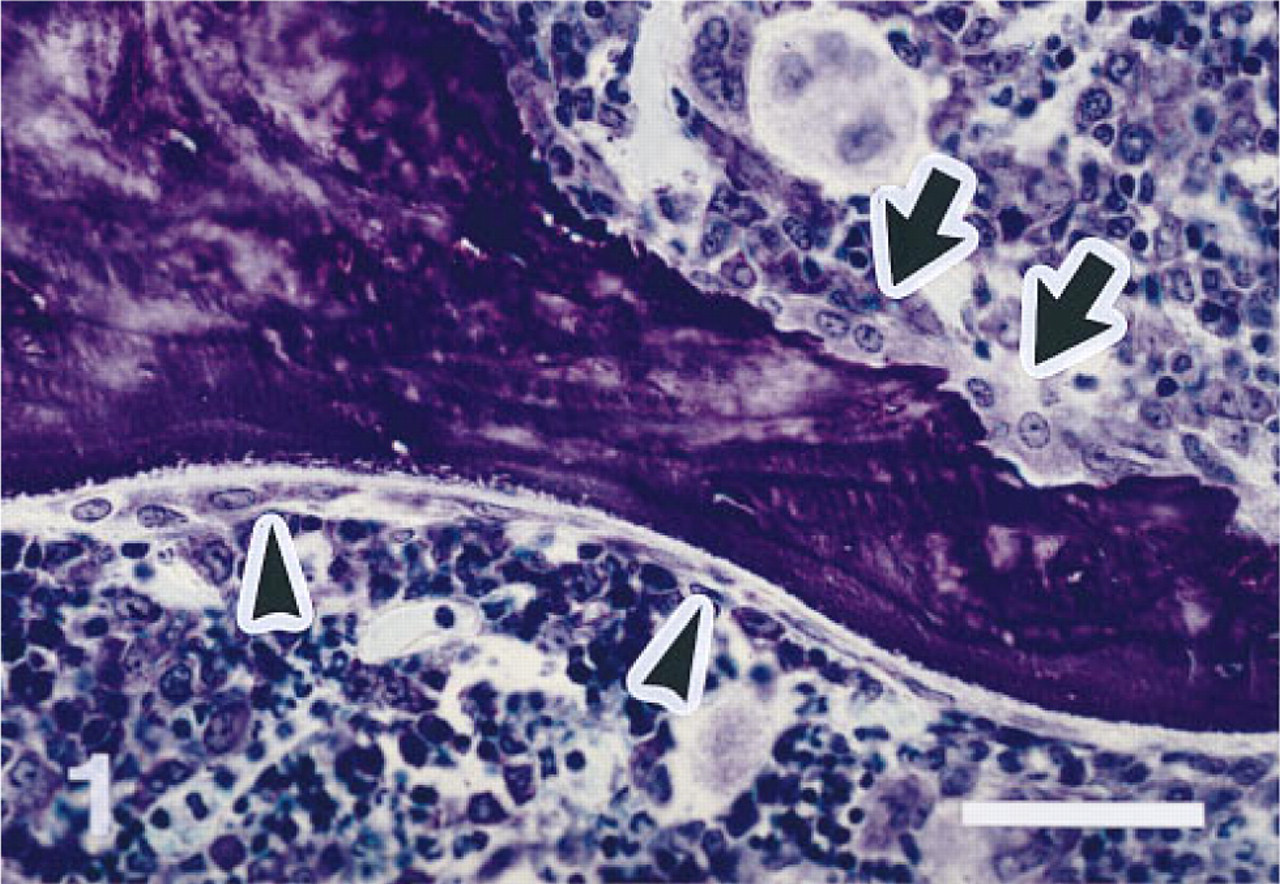
Remodeling sites in vertebral trabecular bone of a 3-month-old female Fischer rat. Bone resorption site with osteoclasts above (arrows), and bone formation site with active osteoblasts on an osteoid seam below (arrowheads). Note good morphological detail, with minimal shrinkage of bone marrow cells. Five-μm-thick undecalcified section of a PFA-fixed and MMA-embedded first lumbar vertebra. Toluidine blue stain. Original magnification X 330. Bar = 50 μm.
When sections of rat liver were exposed to MAb ED1, Kupffer cells in liver sinuses were intensely labeled (Figure 5A). The liver sections were used as positive controls for ED1 labeling. In bone tissue, osteoclasts on periosteal (Figure 5B) and endosteal bone surfaces, as well as osteoclasts (chondroclasts) located beneath the growth plate (Figure 5C), stained strongly positive with MAb ED1. There were also many ED1-positive cells in bone marrow. All cells reactive to ED1 showed a typical cytoplasmic staining pattern (Figures 5A-5C). Reactivity to MAb ED1 in liver and bone tissue was observed with about the same intensity for both ethanol- and PFA-fixed specimens. An isotypespecific non-immune control MAb used at the same concentration as the ED1 antibody did not result in positive staining. At the dilution of primary antibody used in this study, nonspecific background staining was almost absent in sections of liver and bone tissue. Addition of levamisole at the concentration recommended by the manufacturer completely inhibited endogenous AP activity in liver, bone marrow leukocytes, and osteoblasts in most specimens. In ethanol-fixed tissue it was sometimes necessary to double the concentration of levamisole in the incubation medium to inhibit endogenous AP activity.
Discussion
Based on the MMA embedding technique described by Wolf et al. (1992), we have developed an improved method of MMA embedding suitable for histochemical and immunohistochemical investigations in bone tissue as well as for quantitative bone histomorphometry. Compared to the original method, our method is different in several aspects: (a) Instead of dehydration of the bone samples with acetone as described by Wolf et al. (1992), we used a standard dehydration protocol that is also employed by conventional MMA embedding (Schenk et al. 1984). Although acetone can also be used for fixation and dehydration of bone samples, especially for antigens sensitive to formalin or alcohol, we do not recommend its general use because, in our hands, acetone fixation causes massive shrinkage artifacts and acetone dehydration results in inferior quality of the bone section. Using the fixation and dehydration protocol described in this study, our experience, in general, is that antibodies reactive in paraffin-embedded material were also reactive in tissue processed with the MMA embedding technique reported here. (b) The infiltration protocol has been changed to achieve optimal infiltration of the tissue with the plastic. Good infiltration is the basis for uniform polymerization and high section quality. (c) The addition of methylbenzoate to the methacrylate solution during infiltration and polymerization improved preservation of antigenicity of the tissue in our hands. A similar effect has been described for GMA embedding (Britten et al. 1993), and methylbenzoate also increases survival of antigens in paraffin-embedded tissue samples after acetone fixation (Sato et al. 1986). In a similar fashion, the addition of dithiothreitol to all steps of the dehydration, infiltration, and embedding procedure has been reported to considerably improve survival of antigenic determinants in plant tissue embedded in MMA (Baskin et al. 1992). Although the exact nature of the protective effect of these substances during dehydration, infiltration, and polymerization of the plastic is not known, this area certainly deserves further study, because it may be possible to optimize this protective effect in future embedding protocols. (d) Methylbenzoate acts as a plasticizer in MMA embedding mixtures. Uniform polymerization of large MMA-infiltrated specimens can be achieved only when the composition of the last infiltration medium closely resembles the composition of the embedding mixture. Therefore, for the method presented here it was necessary to change the composition of the embedding medium to account for the plasticizing effect of methylbenzoate. (e) The amount of the accelerator N,N-dimethyl-p-toluidine used for chemical polymerization at low temperatures has been reduced in our method, resulting in a lower tendency for bubble formation and more homogeneous polymerization of the blocks. (f) Our method does not require purification and destabilization of the methacrylates used for infiltration and polymerization.
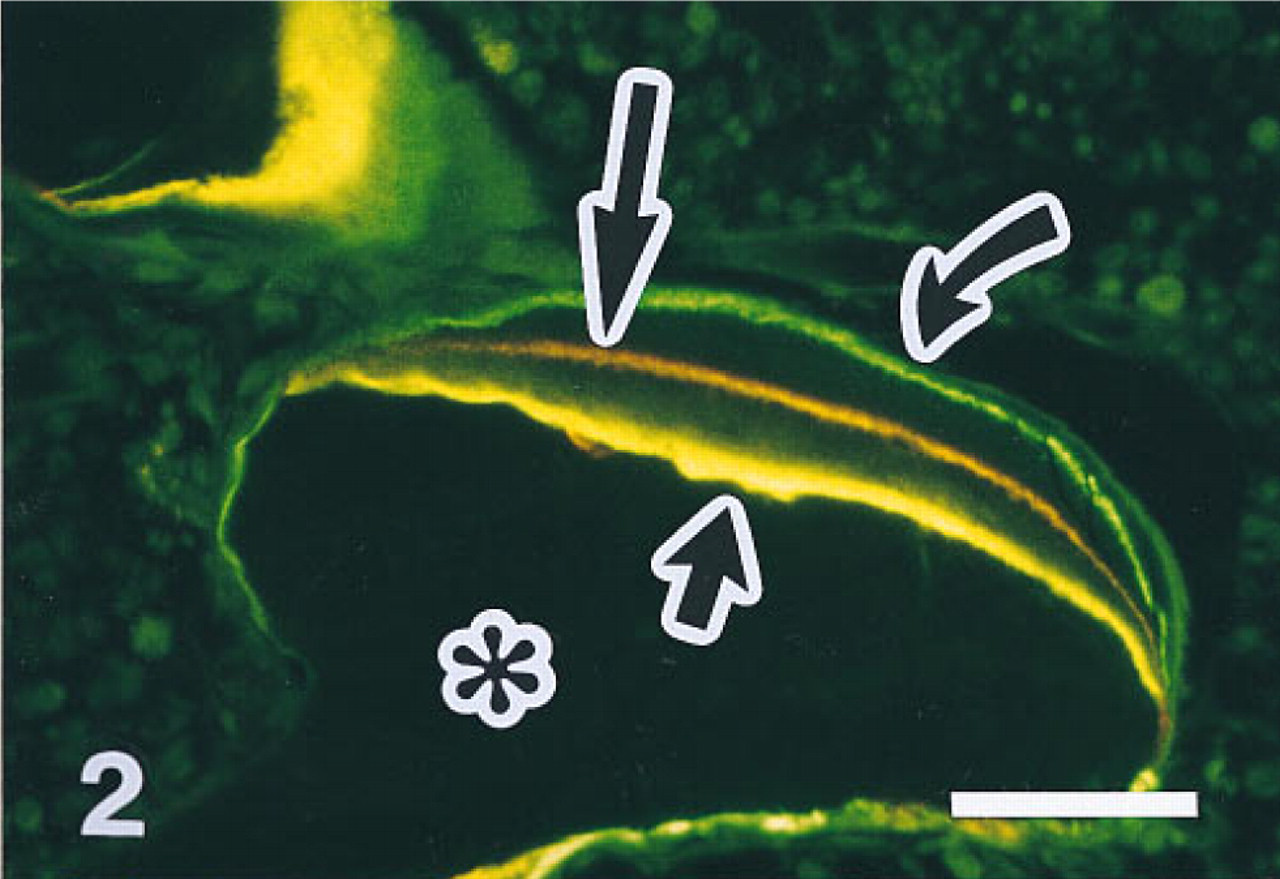
Fluorescent micrograph of vertebral trabecular bone of a 3-month-old rat labeled in vivo with the fluorochromes demeclocycline, alizarin complexone, and calcein. The marker interval between the individual labels was 5 days each. Fluorochrome labeling demonstrates active bone formation in this site (short arrow, demeclocycline; long arrow, alizarin; curved arrow, calcein). Bone appears black (asterisk), and cells in the surrounding bone marrow show some autofluorescence. Five-μm-thick undecalcified section,
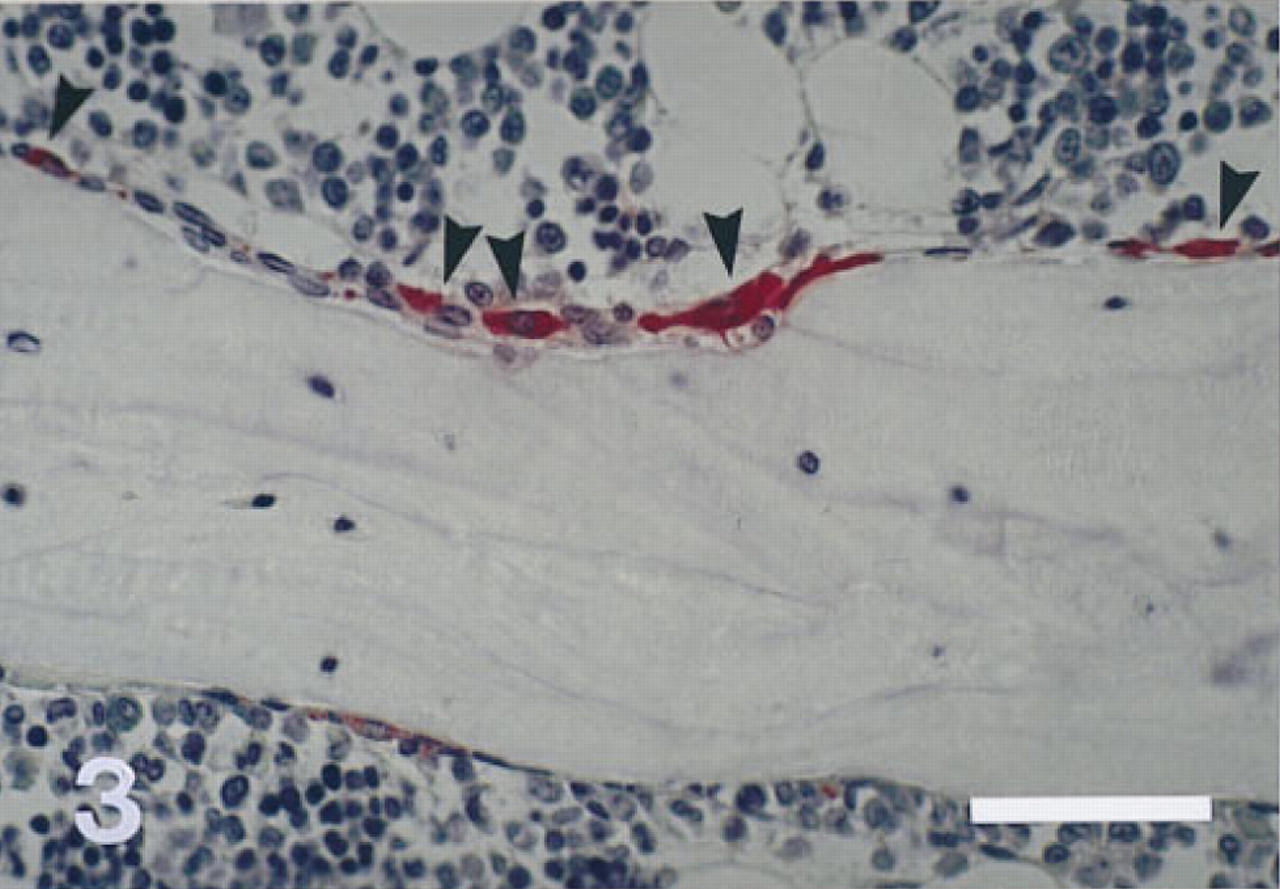
Trabecular bone resorption site with TRAP-positive mononuclear and multinuclear osteoclasts (arrowheads) in a PFA-fixed rat vertebra stained for TRAP and counterstained with Mayer's hematoxylin. Bone marrow cells not in direct contact with the bone surface do not show TRAP activity. Three-μm-thick undecalcified section. Original magnification X 330. Bar = 50 μm.
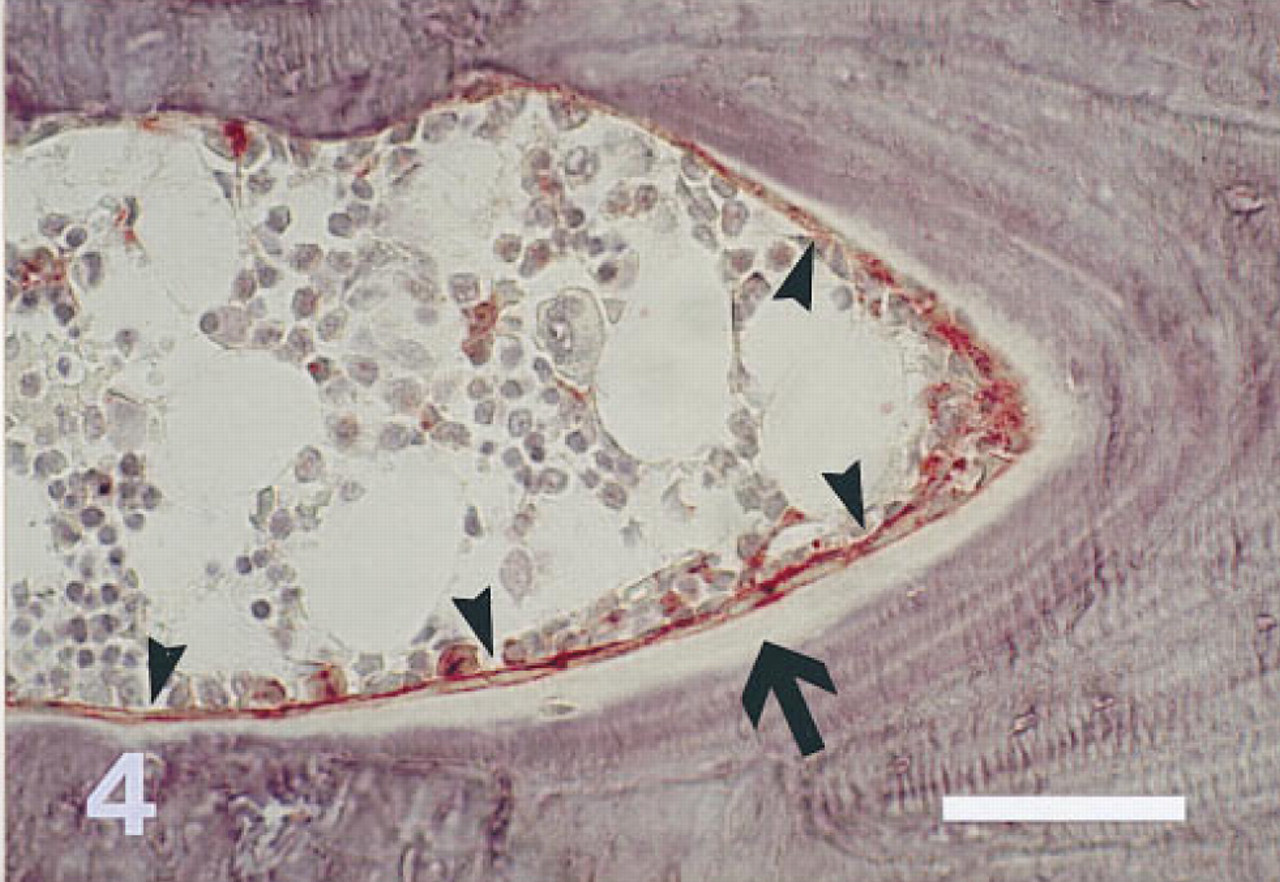
Proximal tibia of a 3-month-old rat fixed in cold 40% ethanol. The section is stained for AP and counterstained with Mayer's hematoxylin. Osteoblasts (arrowheads) in the endocortical bone formation site shown and some bone marrow cells are positive for AP. AP activity is most intense on the apical cell membrane of the osteoblasts. Mineralized bone appears blue-violet; osteoid is marked with an arrow. Three-μm-thick undecalcified section. Original magnification X 330. Bar = 50 μm.
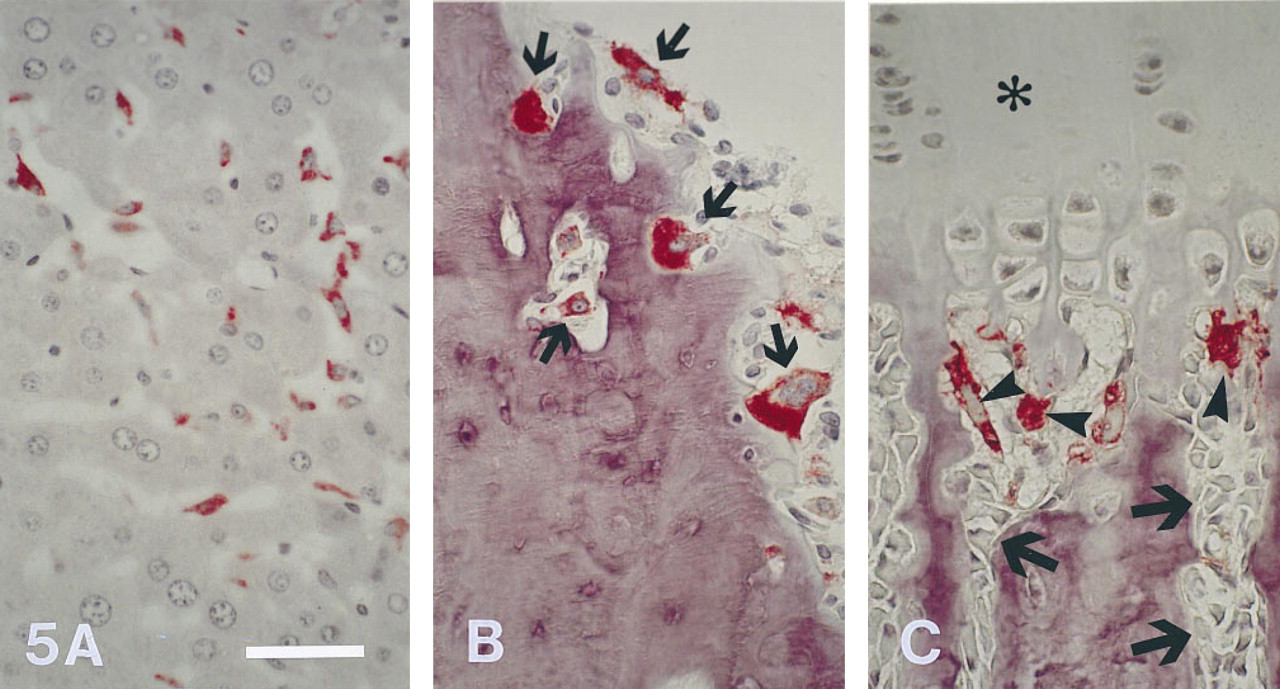
Liver (ethanol-fixed) and tibiae (PFA-fixed) from 3-month-old rats labeled with MAb ED1 (APAAP method). Labeling with ED1 is intense on Kupffer cells in liver sinuses (
High-quality undecalcified sections with few or no artifacts are a prerequisite for reliable histomorphometric measurements (Baron et al. 1983). The embedding technique described here allows the preparation of undecalcified sections with a histological quality comparable to that of conventionally MMA-embedded bone specimens and can therefore be routinely used for bone histomorphometry. We have performed histomorphometry on several hundred bones from rats and other species embedded with this technique in our laboratory. In our experience, the method can be used without modification (except for altered dehydration and infiltration times) for bones of rats, mice, chicken, and cats, as well as for soft tissues. The method may also be applicable to human iliac crest biopsies.
The enzyme activities of AP and TRAP were well preserved by the embedding technique used in this study. TRAP is a lysosomal enzyme expressed at high levels in committed osteoclast precursors and mature osteoclasts (Takahashi et al. 1994). It has recently been shown that TRAP activity in human osteoclasts is resistant to concentrations of tartrate of up to 150-200 mM (Kadoya et al. 1994). At the concentration of tartrate used in this study (100 mM), demonstration of TRAP activity was restricted to osteoclasts and mononuclear cells in close proximity to bone resorption sites in the rat. AP is an enzyme localized on the external surface of the cell membrane of osteoblasts and also odontoblasts (Tenorio et al. 1992; Doty and Schofield, 1984). In agreement with the findings of the present study, it is known that tissue AP activity is very sensitive to fixation (Doty and Schofield 1984).
With the methods employed in this study, Kupffer cells in the liver and osteoclasts were intensely labeled by the MAb ED1. Proteolytic digestion was not necessary for demonstration of positive staining with ED1. In our experience, the rat antigen reacting with MAb ED1 is resistant to most fixatives (e.g., ethanol, acetone, PFA, formalin, Schaffer's solution, and Carnoy's solution). The intense labeling of cells by immunohistological techniques shown in this study is likely due to the fact that complete removal of plastic is possible in sections of MMA-embedded tissue (Baskin et al. 1992). The MAb ED1 recognizes a mainly intracellularly localized 97-kD protein present in almost all cells of the rat mononuclear phagocyte system, and ED1 can be used as a pan-macrophage marker in the rat (Damoiseaux et al. 1994). It is known that rat osteoclasts also stain with MAb ED1 (Lee et al. 1995; Sminia and Dijkstra 1986). The protein that is labeled by MAb ED1 is mainly located on lysosomal membranes and is probably homologous to human CD 68 (Damoiseaux et al. 1994). In frozen sections of bone tissue, human osteoclasts were also shown to stain with an MAb directed against human CD 68 (Kadoya et al. 1994).
In summary, we have described a novel MMA embedding technique that can be used for standard bone histomorphometry and allows the application of histochemical and immunohistochemical methods. Therefore, this method meets the criteria for a more broad application in bone research, and might be useful for the inclusion of histochemical and immunohistological methods in bone histomorphometry. In combination with histochemical or immunohistological methods specific for osteoblasts or osteoclasts, this embedding method may also be useful as a basis for automatic detection of these cells in undecalcified sections of bone tissue by image analysis techniques. Moreover, the embedding technique described here may represent a further step towards the integration of different disciplines into bone research [“molecular histomorphometry” (Parfitt 1994)], and may therefore provide new insights into the physiology and pathology of bone tissue.
Footnotes
Acknowledgements
I thank Stefanie Engert for expert technical assistance.