
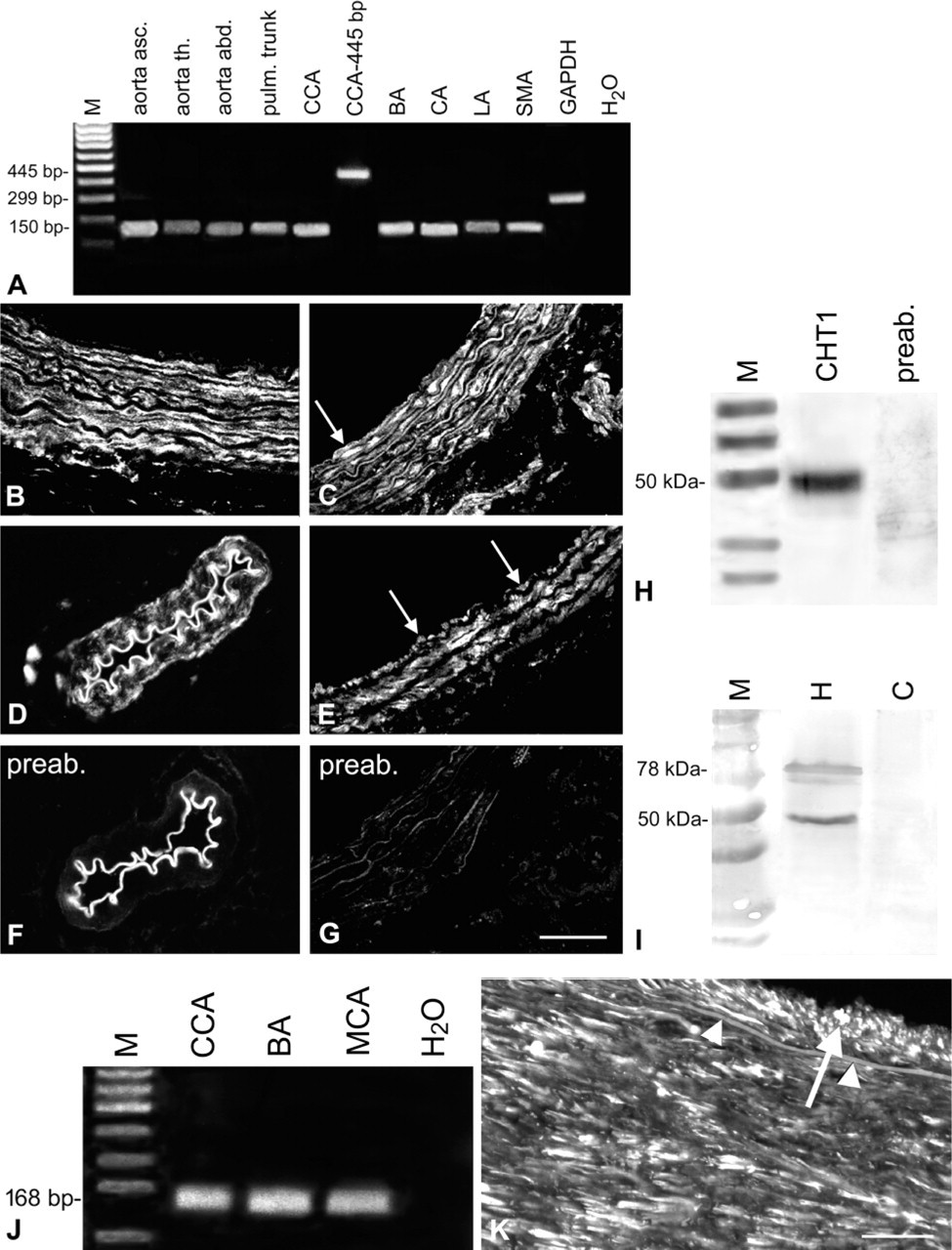
Abstract
The arterial vascular wall contains a non-neuronal intrinsic cholinergic system. The rate-limiting step in acetylcholine (ACh) synthesis is choline uptake. A high-affinity choline transporter, CHT1, has recently been cloned from neural tissue and has been identified in epithelial cholinergic cells. Here we investigated its presence in rat and human arteries and in primary cell cultures of rat vascular cells (endothelial cells, smooth muscle cells, fibro-blasts). CHT1-mRNA was detected in the arterial wall and in all isolated cell types by RT-PCR using five different CHT1-specific primer pairs. Antisera raised against amino acids 29–40 of the rat sequence labeled a single band (50 kD) in Western blots of rat aorta, and an additional higher molecular weight band appeared in the hippocampus. Immunohistochemistry demonstrated CHT1 immunoreactivity in endothelial and smooth muscle cells in situ and in all cultured cell types. A high-affinity [3H]-choline uptake mechanism sharing characteristics with neuronal high-affinity choline uptake, i.e., sensitivity to hemicholinium-3 and dependence on sodium, was demonstrated in rat thoracic aortic segments by microimager autoradiography. Expression of the high-affinity choline transporter CHT1 is a novel component of the intrinsic non-neuronal cholinergic system of the arterial vascular wall, predominantly in the intimal and medial layers.
D
Materials and Methods
RT-PCR
Total RNA was isolated using the RNAzol technique (WAK-Chemie; Bad-Homburg, Germany) from adult Wistar rat (n = 5) ascending, thoracic, and abdominal aorta, pulmonary trunk, superior mesenteric artery, common carotid artery, basilar artery, circulus arteriosus cerebri, lingual artery, from human (n = 3) common carotid artery, basilar artery, medial cerebral artery, posterior cerebral artery, from cultured rat endothelial cells from lung, brain, and aorta, from smooth muscle cells isolated from the common carotid artery, from fibroblasts isolated from lung and from the adventitial layer of the common carotid artery. All studies reported here conformed to NIH guidelines and were approved by the local committees. Samples of human arteries were obtained at autopsy from two male (aged 49 and 78 years) and one female (aged 56 years) subject without evident vascular and neurological diseases. Messenger RNA was isolated from total RNA with oligotex (Qiagen; Hilden, Germany) according to the manufacturer's protocol. Contaminating DNA was destroyed with 1 U DNase/μg total RNA (Gibco–BRL; Karlsruhe, Germany). The mRNA was reverse-transcribed using Superscript RNase H– Reverse Transcriptase (200 U/onset; Gibco-BRL) for 50 min at 42C. For subsequent RT-PCR with the primer pair coding for the 1761-bp CHT1 product (Table 1), 12.5 μl Master Mix (QuantiTect Probe; Qiagen) and 0.6 μl of each primer were supplemented with H2O to a final volume of 25 μl. For RT-PCR with all other primer pairs, 2 μl PCR buffer II, 1.5–2 μl MgCl2 (15 mM), 0.6 μl dNTP (10 mM each), 0.6 μl of each primer (10 μM, Table 1; MWG Biotech, Ebersbach, Germany) and 0.25 μl AmpliTaq Gold polymerase (5 U/μl; Perkin Elmer, Langen, Germany), were supplemented with H2O to a final volume of 25 μl. Cycling conditions were 12 min at 95C, 40 cycles with 45 sec at 95C, 45 sec at 57–65C, 45 sec at 72C, and a final extension at 72C for 7 min. No products occurred in control reactions without template. Primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Table 1) were used as positive controls for efficient RNA isolation and cDNA synthesis. The PCR products were separated by electrophoresis on a 1.2% Tris-acetate-EDTA agarose gel. Sequencing of the PCR products was done by MWG Biotech.
Primer pairs used for RT-PCR
CHT1 mRNA was quantified in samples of rat spinal cord, common carotid artery, and aortic endothelial cells (n = 3 each). Real-time quantitative PCR was done in the iCycler (Bio-Rad; Munich, Germany) using QuantiTec SYBR Green PCR kit (Qiagen). For amplification, the CHT1 primer pair spanning 150 bp (Table 1) was used. The PCR conditions were initial denaturation in one cycle of 15 min at 95C followed by 40 cycles of 30 sec at 95C, 30 sec at 60C, and 30 sec at 72C. All analyses were done in triplicate. The expression of CHT1 was normalized with β2-microglobulin (β2-MG) as a housekeeping gene. The relative expression was calculated by comparison of the received CT values.
Western Blotting
Rat aorta, hippocampus (positive control), and cerebellum (negative control) (n = 5 animals) were lysed in 2 × Laemmli buffer, followed by heating to 65C (10 min). The protein solution was centrifuged for 15 min at 14,000 rpm. An aliquot (5–15 μl) of supernantant was subjected to 10% SDS-PAGE under reducing conditions. The protein was transferred to PVDF membrane (Immobilon-P; Millipore, Bedford, MA) by semi-dry blotting. The membrane was incubated in 25 mM Tris-buffered saline with 0.05% Tween-20 (TTBS, pH 8.0) for 1 hr at room temperature (RT). The affinity-purified CHT1 antibody (Pfeil et al. 2003) was diluted 1:1000 in 5% non-fat dry milk in TTBS and incubated for 12 hr at 4C. Monoclonal alkaline phosphatase-conjugated anti-rabbit IgG from goat (Sigma, Deisenhofen, Germany; 1:15,000 in 2.5% non-fat dry milk in TTBS, 1 hr at RT) was used as secondary antibody and 4-nitroblue tetrazolium chloride-5-bromo-4-chloro-3-indolyl-phosphate (Kirkegaard & Perry Laboratories; Gaithersburg, MD) served as a chromogen. Specificity of immunolabeling was validated by (a) omitting the first antibody and (b) preabsorption of the primary antibody with CHT1 peptide coupled to Sepharose 4B on an affinity column.
Immunofluorescence
Rat (n = 5) ascending, thoracic, and abdominal aorta, pulmonary trunk, superior mesenteric artery, common carotid artery, basilar artery, circulus arteriosus cerebri, lingual artery, and human (n = 5) common carotid artery, basilar artery, medial cerebral artery, and posterior cerebral artery were shock-frozen in melting 2-methylbutane. Samples of human arteries were obtained from the same subjects as described above, and from an additional female (aged 92 years) and male (aged 70 years) subject during dissection at the students' course of macroscopic anatomy. Cryosections were fixed with acetone for 10 min at −20C, and incubated for 1 hr in 10% normal swine serum containing 0.5% Tween-20, 0.1% bovine serum albumin in 0.05 M PBS, pH 7.4. Primary antisera were diluted in PBS (crude anti-CHT1 serum from rabbit 1:16,000, affinity purified anti-CHT1 antibody from rabbit 1:1500, crude anti-CHT1 serum from guinea pig 1:2000), (Lips et al. 2002; Pfeil et al. 2003). Secondary reagents were Cy3-coupled donkey anti-rabbit IgG (1:1000 in PBS; Dianova, Hamburg, Germany), fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG (1:100; Dianova), or biotinylated goat anti-guinea pig IgG (1:50; Sigma); all were applied for 1 hr. Sections incubated with the biotinylated secondary antibody were subsequently labeled with Streptavidin-Texas Red (1:400; Amersham Pharmacia Biotech, Freiburg, Germany). The specificity of immunolabeling was validated by (a) omission the first antibody, (b) incubation with the preimmune serum instead of the primary antiserum, (c) preabsorption of the primary antiserum with CHT1 peptide coupled to Sepharose 4B on an affinity column, and (d) liquid-phase preabsorption with the CHT1 peptide (10–200 μg/ml) that was used for immunization. Sections were rinsed, coverslipped with carbonate-buffered glycerol (pH 8.6), and evaluated with an epifluorescence microscope (Axioplan 2; Zeiss, Oberkochen, Germany) and a confocal laser scanning microscope (Leica TCSSP; Mannheim, Germany).
Cell Culture
Microvascular endothelial cells of forebrain and lung and of rat aorta (n = 3) were isolated by a magnetic bead (Dyna-bead)-coupled endothelial cell-specific antibody (anti-RECA; Serotec, Düsseldorf, Germany). For this purpose, the RECA antibody was diluted 1:20 in 0.05 M PBS and incubated with a 5% solution of 4 × 108 Dynabeads pan mouse IgG (Dynal Biotech; Hamburg, Germany) for 16 hr at 4C. Adult Wistar rats were sacrificed by inhalation of sevofluran (Abbott; Wiesbaden, Germany). Samples were minced and incubated end over end with Dynabead-coupled anti-RECA solution for 4 hr at RT. Endothelial cells coupled with the Dynabead-conjugated RECA antibody were separated by using a magnet and suspended in Endothelial Cell Growth Medium-MV (Promocell; Heidelberg, Germany). The cells were characterized by their binding capacity for the isolectin-B4 (biotin-labeled I-B4 lectin, 1:50, Sigma, followed by Streptavidin–TexasRed 1:400, Amersham, Braunschweig, Germany) (Porter et al. 1990), expression of CD31 (mouse monoclonal antibody MEC13.3, 1:100; Pharmingen, Hamburg, Germany, followed by donkey anti-mouse IgG, Cy3 conjugate, 1:1000, Dianova) and α-smooth muscle actin (FITC-conjugated monoclonal antibody 1A4, 1:500; Sigma) (Muzykantov et al. 1999; Jones et al. 2000), and by uptake of acetylated LDL (DiI-conjugated Ac-LDL; Paesel and Lorei, Hanau, Germany) applied to the culture at 10 μg/ml for 4 hr) (Craig et al. 1998). More than 90% of the cells were I-B4-positive, showed immunoreactivity for CD31 but not for α-smooth muscle actin, and contained acetylated LDL.
Vascular smooth muscle cells were isolated from rat common carotid artery. The endothelium and the adventitial layer of the artery were abraded before incubation in 1 ml 0.05 M PBS with 2.5 mg collagenase CLS II (Biochrom AG; Berlin, Germany), 200 μl 0.1% trypsin/0.05% EDTA (PAA; Cölbe, Germany), and 50 μg DNase I (Boehringer, Ingelheim, Germany) for 2 hr at 37C. The pellet was washed twice with PBS/DNase I, and the cells cultured to confluence in RPMI 1640 tissue culture medium (Gibco-BRL) supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin/amphotericin at 37C. To remove remaining fibroblasts, cells were detached with 0.1% trypsin/EDTA and incubated with 500 μl fibroblast-specific antibody solution (1:100, F4771; Sigma) and 25 μl Dynabeads for 1 hr at 37C. Vascular smooth muscle cells were washed twice and resuspended with 1640 RPMI medium with 10% fetal bovine serum and 1% penicillin/streptomycin/amphotericin. The purity of these cultures was verified by immunolabeling for α-smooth muscle actin. Roughly 1% of cells remained unlabeled with this antibody.
For isolation of fibroblasts, common carotid artery and lung were incubated for 30 min in 1 ml 0.05 M PBS with 100 μl 0.1% trypsin/EDTA and 2.5 mg collagenase CLS II, and for 90 min in 1 ml PBS with 200 μl trypsin and 10 μg DNase I at 37C. The pellet was washed twice with PBS/DNase I and the cells cultured to confluence in RPMI 1640 tissue culture medium supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin/amphotericin at 37C. For IHC, the cells were detached with 0.1% trypsin/EDTA and resuspended in RPMI 1640 medium containing 10% fetal bovine serum and 1% penicillin/streptomycin/amphotericin. Five thousand cells per well were grown in eight-well chamber slides. After 2 days, cells were fixed in phosphate-buffered 4% paraformaldehyde solution for 20 min, followed by washing steps and incubation with primary and secondary antisera as described above for tissue sections.
In Vitro [3H]-choline Uptake
Rat thoracic aorta (n = 3) was dissected into three specimens and washed in NaCl-free buffer containing 5 mM Tris, 10 mM Hepes, 2 mM KCl, 1 mM MgCl2, and 1 mM CaCl2 (pH 7.4). The first specimen was incubated in 200 nM [3H] -choline (545 mCi/mg; Amersham Pharmacia Biotech) diluted in the same buffer as used for washing but with 200 mM NaCl. The second specimen was incubated in [3H]-choline-containing buffer without NaCl, and the third in [3H]-choline-containing buffer with 50 μM hemicholinium-3 (Sigma) for 10 min. After washing, the tissue was frozen and cryosections (20 μm) were prepared, dried, and analyzed for 24 hr with a Microimager (Zinsser Analytic; Frankfurt/Main, Germany).
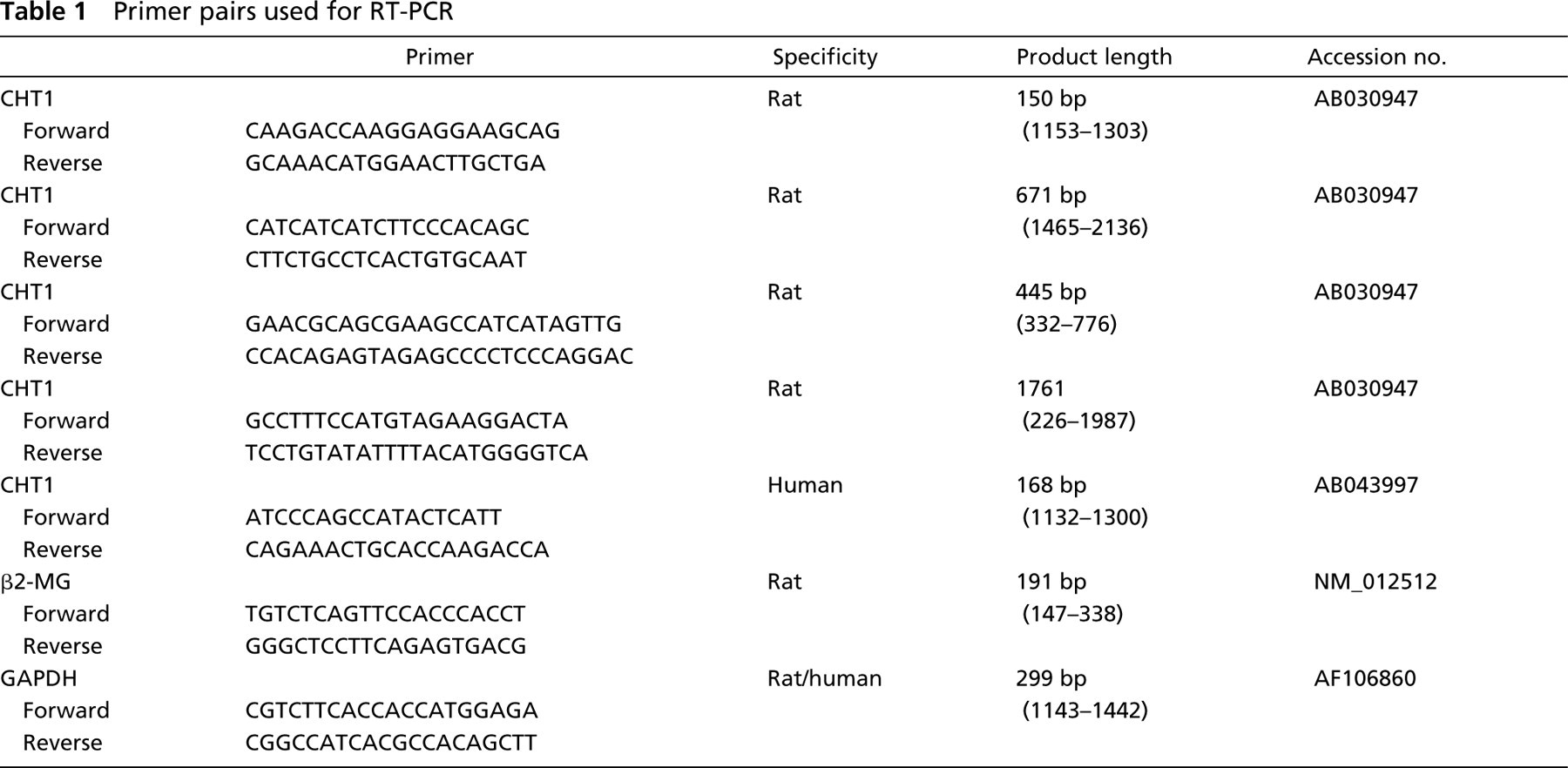
CHT1 expression in cultured vascular cells. (
Results
CHT1 Expression in Cultured Vascular Cells
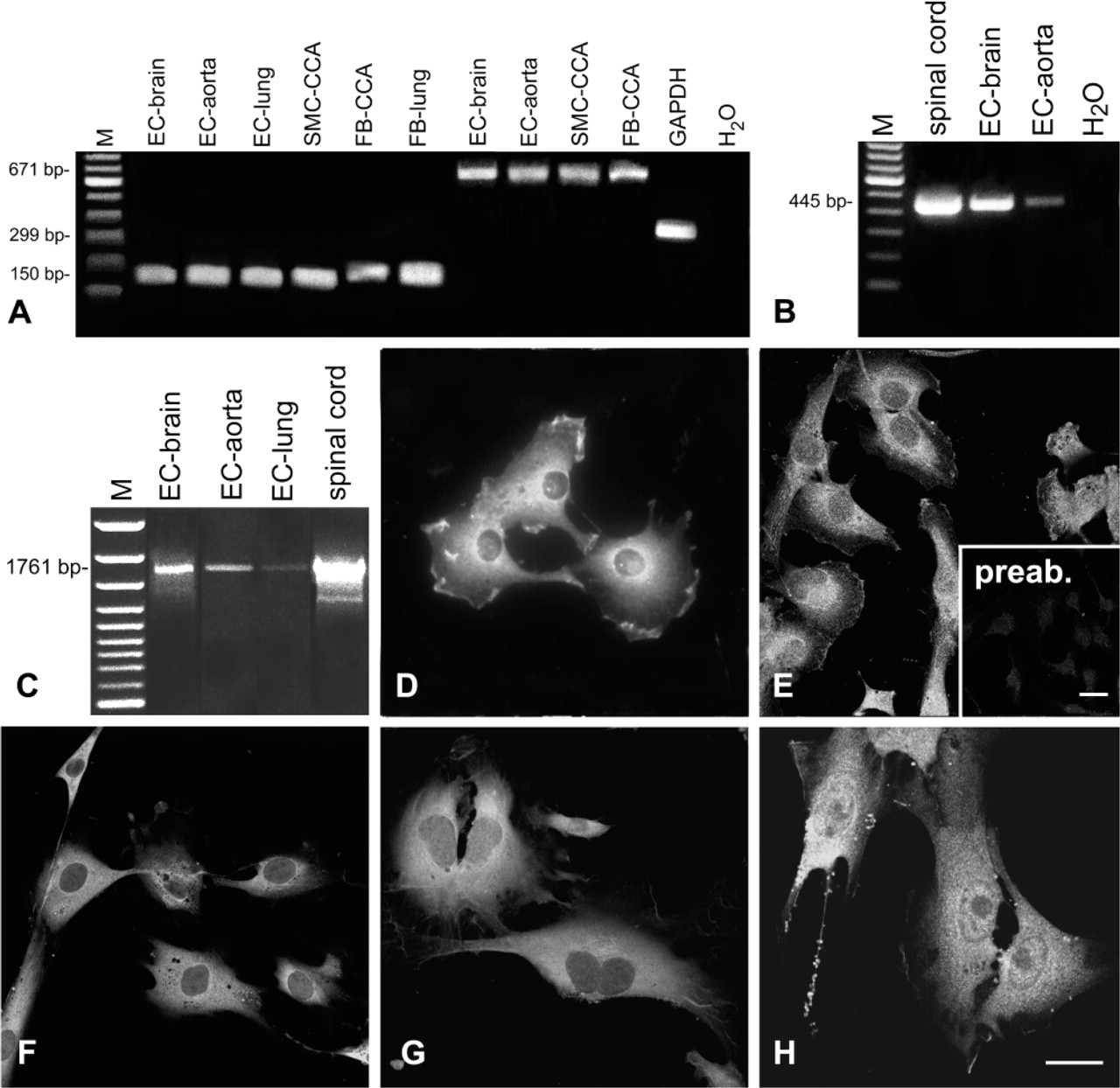
CHT1 mRNA was detected in cultured vascular cells using all four different rat-specific primer pairs (Table 1; Figures 1A–1C). Using primer pairs coding for a 150-bp and a 671-bp product, CHT1 mRNA was detected in samples of the endothelial cells from brain, lung, and aorta, of the smooth muscle cells of the common carotid artery, and of fibroblasts from lung and common carotid artery (Figure 1A). With the gene-specific primer pair first used by Friedrich et al. (2001), we amplified a CHT1 product of the appropriate length of 445 bp in endothelial cells of aorta and in microvascular endothelial cells of brain and lung (Figure 1B). Products obtained with all three primer pairs were sequenced and found to be identical to the sequence known from cholinergic neurons. In the same samples the complete coding sequence was amplified with a primer pair for a 1761-bp CHT1 product (Figure 1C). Quantitatively, CHT1 expression in the rat common carotid artery and aortic endothelial cells amounted to 26% and 29%, respectively, of that in spinal cord, as revealed by real-time RT-PCR (Figure 2). Endothelial cells, smooth muscle cells, and fibroblasts cultured from all sources exhibited CHT1 immunoreactivity. CHT1 immunolabeling was observed both in the membrane and intracellularly in a granular pattern, predominantly surrounding the nucleus (Figures 1D–1H). Some cells displayed CHT1-immunoreactive processes (Figure 1H).
CHT1 Expression in Rat and Human Arteries
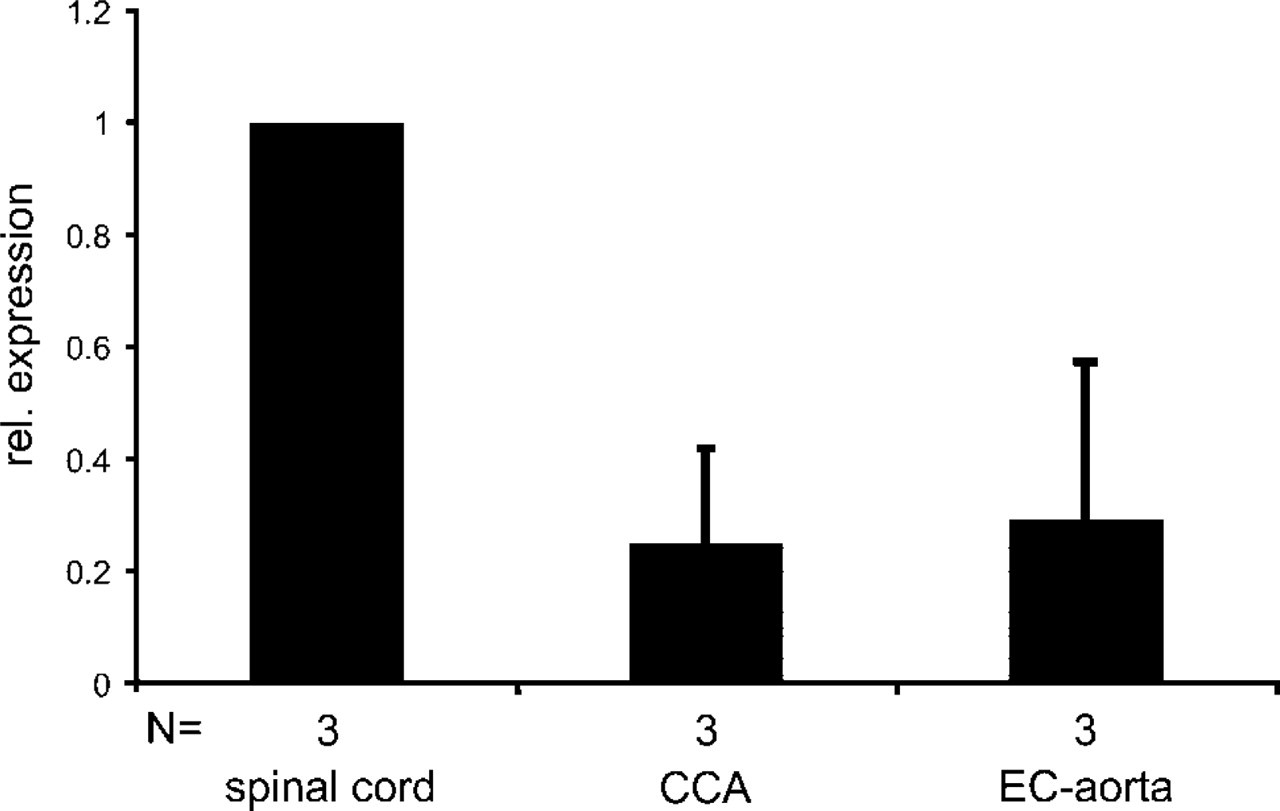
Messenger RNA coding for CHT1 was detected in all investigated rat specimens with the CHT1 primer pair amplifying a PCR product of 150 bp (Figure 3A) and verified by sequencing. Using the CHT1 primer pair described by Friedrich et al. (2001), a product of 445 bp in length was obtained in the common carotid artery (Figure 3A). In Western blots, a single band of 50 kD was detected in homogenates of the rat aorta. Immunolabeling of this band was abolished after preabsorption of the primary antiserum with CHT1 peptide coupled to a Sepharose 4B affinity column (Figure 3H). In homogenates of the rat hippocampus, a band of 50 kD and a weaker band at 78 kD were observed in Western blots, while no immunoreactive protein was detected in rat cerebellum that served as negative control (Figure 3I). IHC showed the presence of CHT1-IR in smooth muscle cells of all investigated rat arteries (Figures 3B–3G). Endothelial cells were consistently immunolabeled in the pulmonary trunk (Figure 3E) and endothelial CHT1 immunoreactivity was inconsistently found in other vessels, with lowest occurrence in the lingual artery (Figures 3B–3E). Labeling of adventitial fibroblasts was sparse. Nerve fiber bundles accompanying the lingual and superior mesenteric artery were strongly stained with the CHT1 antibody (Figures 3C and 3D).
Relative expression (mean ± SD) of CHT1 mRNA in rat common carotid artery (CCA) and aortic endothelial cells (EC-aorta) in comparison to spinal cord, as determined by quantitative RT-PCR.
Products corresponding to CHT1 mRNA were also detected in all investigated human arteries with primers coding for a 168-bp product (Figure 3J). The identity to the sequence known from neural tissue was verified by sequencing. IHC showed strong CHT1-IR in smooth muscle cells and in cells of the subendothelial layer (Figure 3K).
[3H]-Choline Uptake by Aortic Rings
Specimens of the rat thoracic aorta were used to investigate the [3H]-choline uptake in vitro. In the presence of 200 mM NaCl in the incubation buffer, [3H]-choline uptake was prominent in the medial layer (Figure 4A). The spatial resolution of the autoradiographic technique (∼20 μm) did not allow us to discriminate the intimal layer unequivocally from the media. Spots of [3H]-choline uptake were inconsistently observed in the adventitial layer. Specimens incubated in [3H]-choline buffer without NaCl displayed a weak signal throughout the vascular wall (Figure 4B). When specimens were incubated with [3H]-choline in the presence of hemicholinium-3, incorporation of radioactivity was reduced to a minimum (Figure 4C).
Discussion
The present findings provide several lines of evidence for the expression of the high-affinity choline transporter CHT1, which was originally been identified in nervous tissue (Okuda et al. 2000), in various layers of the arterial wall. At the transcriptional level, CHT1-specific products could be amplified by RT-PCR using five different primer pairs, including one spanning the entire coding region. At the translational level, two antisera raised against amino acid residues 29–40 of the rat CHT1 labeled cells of the arterial wall in situ as well as in culture conditions after isolation. With the same antisera, a single 50-kD protein was detected by Western blotting analysis in the rat aorta, which lacks autonomic innervation (Morris and Gibbins 1990), while an additional band of 78 kD appeared in the hippocampus. The band of higher apparent molecular weight (78 kD) exceeds that predicted from the cloned rat sequence, i.e., 63.4 kD (Okuda et al. 2000) and might reflect a glycosylated variant. The 50-kD band corresponds well with that of rat CHT1 expressed by transfected cells (Okuda et al. 2002) and with CHT1-immunoreactive protein from the tracheal epithelium as determined by Western blotting (Pfeil et al. 2003). These data suggest a tissue-specific processing of CHT1 in that non-neuronal CHT1 transfected cells as well as physiologically CHT1-expressing non-neuronal cells process CHT1 to a 50-kD protein, while an additional higher molecular weight protein is generated only in the nervous system. Both CHT1 mRNA and CHT1 immunoreactivity were detected in endothelial cells, smooth muscle cells, and adventitial fibroblasts. Endothelial cells of various origins have long been identified as cholinergic cells displaying ChAT immunoreactivity (Parnavelas et al. 1985; Haberberger et al. 2000), ChAT activity (Gonzalez and Santo-Benito 1987), expressing VAChT (Haberberger et al. 2000; Kirkpatrick et al. 2001), and releasing ACh (Milner et al. 1989; Kawashima et al. 1990). Hence, an efficient uptake system for choline that reaches beyond the low-affinity choline transporters that are required by any cell for uptake of choline destined for membrane phospholipid synthesis would be expected in these cells. Moreover, brain capillary endothelial cells have to shuffle choline across the blood–brain barrier to make it available for neuronal ACh synthesis in the central nervous system. Consequently, choline uptake by endothelial cells has received attention and has been studied in several models with particular focus on the blood–brain barrier. Recent detailed studies were conducted on cell lines serving as models for brain microvascular endothelial cells. Both the murine cell line MBEC4 and the rat cell line RBE4 express a high-affinity choline uptake system that does not fully match the functional characteristics of CHT1 (Sawada et al. 1999; Murakami et al. 2000; Friedrich et al. 2001), and no RT-PCR product could be amplified from RBE4 cells using a primer pair spanning bp 332–776 of rat CHT1 (Friedrich et al. 2001). In contrast, we obtained CHT1-specific signals in primary cell cultures of rat brain endothelial cells harvested by immunomagnetic isolation both in RT-PCR using four different primer pairs and in IHC using two different antisera. This suggests that endothelial cells in situ and in primary culture may differ in this aspect from cell lines. This assumption is supported by the presence of ChAT activity in endothelial cells isolated from rat brain cortex capillaries (Gonzalez and Santo-Benito 1987) but its lack in RBE4 cells (Malo et al. 1999).
CHT1 expression in rat (
Nevertheless, choline uptake kinetics (Km, Vmax) differing from those of neuronal CHT1 have also been reported using the in situ rat brain perfusion technique (Allen and Smith 2001) and in studies using isolated bovine brain capillaries (Galea and Estrada 1992) and endothelial cells (Estrada et al. 1990). Sodium dependence, one of the characteristics of neuronal CHT1 (Haga 1971), has been reported in some of the studies (Estrada et al. 1990; Galea and Estrada 1992) but not in others (Allen and Smith 2001). Sensitivity of choline uptake to hemicholinium-3, a key feature of neuronal CHT1 (Chang and Lee 1970), has been observed in all types of studies, including endothelial cell lines and extracerebral, i.e., aortic endothelial cells, but with slightly different inhibition constants (Allen and Smith 2001; Friedrich et al. 2001; Lipton et al. 1988). The reasons for the discrepancies between the expected functional characteristics due to the presence of CHT1 mRNA and protein (this study) and those observed in previous functional studies remain to be determined. Because even a single nucleotide polymorphism in human CHT1 can have a profound impact on choline uptake (Okuda et al. 2002), one, but not necessarily the only, possible explanation is offered by cell type-specific modifications of CHT1, as suggested by the Western blotting data, or by association with different modifying proteins.
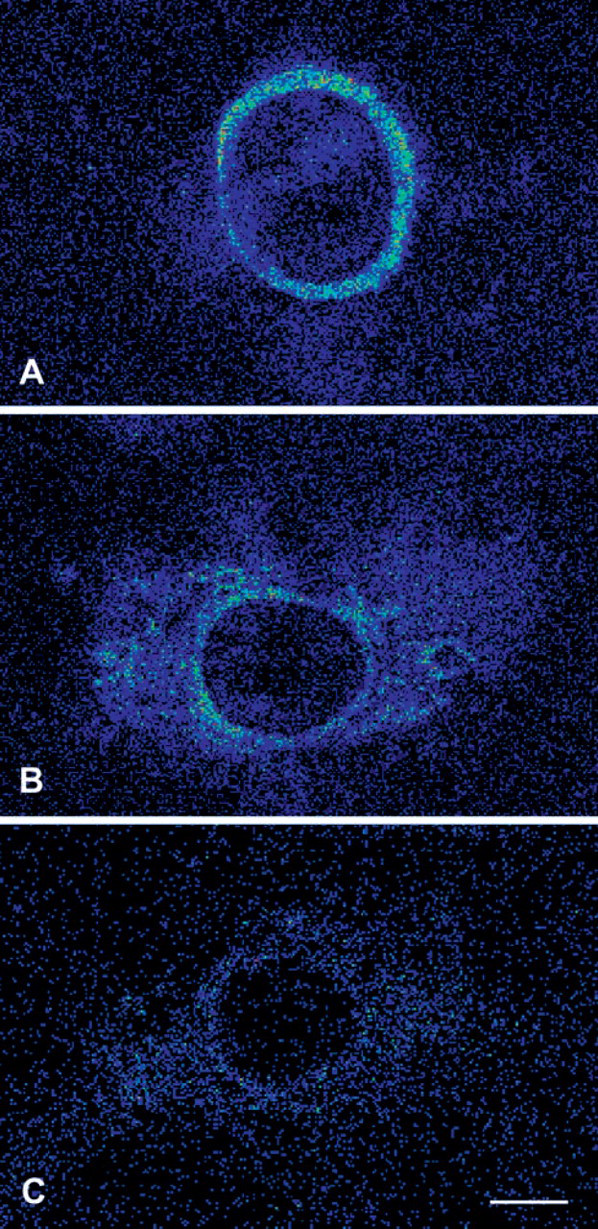
Autoradiographic (Microimager) demonstration of [3H]-choline uptake. (
The strongest immunolabeling for CHT1 in the arterial wall was observed in smooth muscle cells of the tunica media. Cultured vascular smooth muscle cells expressed CHT1 mRNA as determined by RT-PCR and also exhibited CHT1 immunoreactivity. At first sight these findings might appear surprising because smooth muscle cells are not classically considered as being cholinergic cells. However, there are several reports documenting the ability of muscle cells to synthesize ACh, either via ChAT or via the enzyme carnitine acetyltransferase. ACh-synthesizing activity and/or ACh release have been demonstrated in myo-blasts (Krause et al. 1995; Fu et al. 1998), skeletal muscle fiber (Tucek 1982; Miledi et al. 1982), and smooth muscle fibers of the human skin (Wessler and Kirkpatrick 2001). Moreover, airway smooth muscle cells have been shown to display ChAT immunoreactivity (Wessler and Kirkpatrick 2001). In support of CHT1 expression in vascular smooth muscle cells, [3H]-choline uptake was prominent in the aortic tunica media, as demonstrated by autoradiography in the present study, and this uptake shared crucial characteristics with neuronal CHT1, i.e., sensitivity to hemicholinium-3 and dependence on sodium. Hence, there is ample evidence for CHT1 expression in vascular smooth muscle cells, and further detailed kinetic studies will be required to elucidate the extent to which their high-affinity choline uptake matches that of the nervous system.
In culture, fibroblasts prepared from the common carotid artery and from the lung also exhibited distinct CHT1 immunoreactivity and expressed CHT1 mRNA. Fibroblasts and myofibroblasts are well-established targets of ACh, carrying both muscarinic and nicotinic ACh receptors (Andre et al. 1988; Sekhon et al. 2002; Jacobi et al. 2002; Oben et al. 2003). Evidence for ACh synthesis by fibroblasts, however, is sparse and as yet has been presented only in abstract form for lung fibroblasts (Proskocil et al. 2000) while several other investigators measured ACh release from fibroblasts only after transfection with ChAT (Fisher et al. 1993; Falk–Vairant et al. 1996). These findings fit best with the assumption that fibro-blast ACh synthesis may be of low level in general and depends in its extent on culture conditions. Consistent with this notion, CHT1 immunolabeling of adventitial fibroblasts in the intact arterial wall and [3H]-choline uptake in the adventitial layer were inconsistent and of low labeling intensity. Moreover, adventitial [3H] -choline uptake cannot be attributed to fibroblasts with certainty because a clear discrimination between vasa vasorum, small nerve fiber bundles, and fibro-blasts was not possible with by the present autoradiographic technique.
In conclusion, the present data show that expression of the high-affinity choline transporter CHT1 is a novel component of the intrinsic non-neuronal cholinergic system of the arterial vascular wall, predominantly in the intimal and medial layers.
Footnotes
Acknowledgements
Supported by HMWK (to KSL) and DFG (SFB 547, project A7 to RCBD), and project C2 to (WK and RVH).
We thank Ms S. Tasch, Ms K. Michael, Mr M. Boden-benner, and Mr G. Weigand for skillful technical assistance, and Ms Ch. Becker (Institute for Pathology, Justus-Liebig-University Giessen) for her help in collecting the human samples.