
Abstract
In spite of intensive investigations, the roles of acetylcholinesterase (AChE; EC 3.1.1.7) and butyrylcholinesterase (BuChE; EC 3.1.1.8) in the central nervous system (CNS) remain unclear. A role recently proposed for BuChE as an explanation for survival of AChE knockout mice is compensation for AChE activity if it becomes insufficient. Neuronal contribution of both enzymes to the cholinesterase pool in the neuromuscular junction has also been suggested. These proposals imply that BuChE expression follows that of AChE and that, in addition to AChE, BuChE is also expressed in α-motor neurons. However, these assumptions have not yet been properly tested. Histochemical approaches to these problems have been hampered by a number of problems that prevent unambiguous interpretation of results. In situ hybridization (ISH) of mRNAs encoding AChE and BuChE, which is the state-of-the-art approach, has not yet been done. Here we describe rapid nonradioactive ISH for the localization of mRNAs encoding AChE and BuChE. Various probes and experimental conditions had been tested to obtain reliable localization. In combination with RT-PCR, ISH revealed that, in rat spinal cord, cells expressing AChE mRNA also express BuChE mRNA but in smaller quantities. α-Motor neurons had the highest levels of both mRNAs. Virtual absence of transcripts encoding AChE and BuChE in glia might reflect a discrepancy between mRNA and enzyme levels previously reported for cholinesterases.
Keywords
P
Gene-targeted mice with totally eliminated AChE activity have been developed recently (Xie et al. 2000). These mice were able to live to adulthood (Li et al. 2003), although it is known that AChE inhibitors are highly lethal and cause death of wild-type mice in a few minutes (Heath 1961; Taylor 1996). Increased sensitivity to BuChE-specific inhibitors led to the explanation that BuChE activity, which remains normal in AChE knockout mice, enables their survival and that the role of BuChE might be to compensate for the function of AChE if it is for any reason compromised (Li et al. 2000; Xie et al. 2000). However, this explanation failed to be proved in mutant mice deficient in the collagen tail-like structural subunit (ColQ) that serves to bind the asymmetric A12 AChE molecular form to the specialized basal lamina of the neuromuscular junction (NMJ). Specifically, BuChE inhibitors did not cause significant change in decay time of the miniature end plate potentials, and the authors therefore abandoned the explanation that BuChE activity compensates for the absence of AChE activity in these functionally normal NMJs (Feng et al. 1999). Biochemically, they demonstrated membrane-associated BuChE but were not able to specify whether it is associated with nerve terminals or with terminal Schwann cells in the NMJs (Feng et al. 1999).
The questions and dilemmas concerning the roles of AChE and BuChE could not be solved without identification of cells expressing these two enzymes. Assuming that BuChE is capable of playing a back-up role for AChE, one would expect related expression of the two enzymes. It is therefore important to find out whether or not the cells expressing AChE also express BuChE. Moreover, the proposed presynaptic location of BuChE in the NMJ (Feng et al. 1999) requires that it be expressed in motor neurons, which has not yet been established.
Several studies have previously approached AChE and BuChE localization in the CNS. They have relied either on biochemical methods, using homogenization and enzyme isolation, or the more direct histochemical approach (reviewed in Silver 1974; Darvesh et al. 2003). However, these techniques are not suitable for identification of cells expressing cholinesterases in the CNS. Mature enzymes can either be released into the surrounding space or transported along dendrites and axons far away from the perikarya of their origin, and the cell bodies containing AChE are masked by an intensely labeled neuropil. Investigators tried to overcome this problem by irreversible inhibition of the preexisting enzyme, followed by enzyme staining shortly after inhibition. In this way only de novo synthesized enzyme, still confined to the perikarya of the expressing cells, is visualized (Woolf and Butcher 1986). However, a higher ACh concentration in the region resulting from enzyme inhibition and other indirect effects of anticholinesterases might influence cholinesterase expression so that unambiguous identification of cell populations producing AChE and BuChE under normal conditions cannot be achieved in this way. In addition, only catalytically active enzymes are detected by the thiocholine technique, leaving the pool of inactive AChE (Massoulié et al. 1996) undetected.
Here we identified the cells expressing AChE and BuChE in the rat spinal cord by ISH of their mRNAs. This is the method of choice for such investigations because mRNAs remain confined to the perikarya of their origin and are therefore reliable markers for the cells that express the enzymes. Nonradioactive digoxigenin labeling of the probe was selected. To determine the optimal conditions for localization of mRNAs encoding AChE and BuChE, we first tested several variants of prehybridization and hybridization steps. We compared oligonucleotide and RNA probes and tested them in the rat striatum, where AChE mRNA has a very specific and well-recognizable distribution, as confirmed in various labs by different in situ approaches (Landwehrmeyer et al. 1993; Hammond et al. 1994; Bernard et al. 1995). For localization of AChE mRNA, we used two probes, one from the second exon that recognizes all AChE mRNA species and another probe that was selective for the tailed (T) AChE mRNA variant. The number of cells and their identity in the tissue sections were determined by fluorescent nuclear staining. Cholinergic neurons were visualized by choline acetyltransferase (ChAT) staining.
Materials and Methods
Animals
Female adult wistar rats (180–200 g) were sacrificed by exposure to CO2 narcosis. The cage was covered and CO2 was applied at a rate that displaced at least 20% of the chamber volume per minute until death, as specified by the Veterinary Administration of the Ministry for Agriculture, Forestry and Food of Slovenia (permit No: 323–02–74/00). All efforts were made to minimize the number of animals used and their suffering. For ISH of AChE mRNA and BuChE mRNA and for immunocytochemistry of ChAT, the thoracic spinal cord was lifted out on ice, frozen on dry ice, and stored at −80C until ready for sectioning. Cross-sections of the frozen thoracic spinal cord 25 μm thick were prepared with a cryostat at −20C and mounted on Probe On+ slides purchased from Fisher Scientific (Pittsburgh, PA). Sections were kept in a dry air chamber at −20C until use.
Probes for ISH
RNA Probes for AChE mRNA Localization. DIG-11 UTPs (Boehringer-Roche; Mannheim, Germany) were used for nonradioactive labeling of RNA probes by digoxigenin. Two probes were used for the AChE mRNA localization, here referred to as AChE 1 and AChE 2. AChE 1 was transcribed from the pGEM 3z vector and corresponded to a 339-bp AChE gene fragment (nucleotides 370–709, rat sequence) from the second exon. This probe was therefore directed to all AChE mRNA species present in the rat CNS. Probe AChE 2 was transcribed from the pGEM T-easy vector and corresponded to a 332-bp AChE gene fragment from exons 4 and 6 (nucleotides 1471–1801, rat sequence). This probe was therefore specific for the tailed (T) AChE mRNA species. In the control experiments we replaced antisense DIG-labeled RNA probes with sense probes that were DIG-labeled and transcribed from the same vector using an alternative promoter site.
Oligonucleotide Probe for AChE mRNA Localization. For the AChE mRNA localization we also tested non-radioactively labeled oligonucleotide probes. The sequence of synthesized oligonucleotides (MWG-Biotech; Ebersberg, Germany) was the same as used for the AChE mRNA localization by Hammond et al. (1994) and corresponded to nts 1090–1152 (rat sequence). Instead of [33P]-dATP, used for 3′ labeling in the original study, we labeled our oligonucleotides at the same 3′ position with digoxigenin-dUTP according to the producer's manual (DIG Oligonucleotide Tailing Kit; Boehringer-Roche). A 40-nt-long tail that includes four digoxigenin-tagged nucleotides is predicted at the 3′ location at a concentration ratio DIG-dUTP:dATP of 1:10.
RNA Probe for BuChE mRNA Localization. This probe corresponded to the 353-bp BuChE gene fragment located in exons 2, 3, and 4 (nts 1446–1798, rat sequence). In preparation of this probe we were careful to select the sequence that had minimal homology with the AChE cDNA. The selected sequence was therefore aligned with all known rat cDNA sequences with the aid of a BLAST computer program. No homology to any other rat cDNA, including AChE cDNA, was found according to the standard criteria of the BLAST program. In this way we prevented any binding of the BuChE probe to the AChE mRNA at the stringency employed in the BuChE mRNA ISH. RNA antisense, and control sense probes were labeled and prepared from the pGEM T-easy vector in the same way as described above.
ISH Procedure
The tissue sections were fixed for 5 min with 4% formaldehyde. After rinsing in PBS and 70% ethanol, sections were kept in 95% ethanol. To test the efficiency of the acetylation reaction in reducing the nonspecific binding of the probe, we compared AChE mRNA localizations obtained with and without the acetylation step in the prehybridization stage. For acetylation, sections were washed with PBS, incubated for 10 min in freshly prepared acetic anhydride solution (0.25 vol% acetic anhydride, 0.1 M triethanolamine-HCl, 0.9% NaCl, pH 8.0) and then rinsed first in 70% and then in 95% ethanol. During prehybridization, samples were kept in a humid chamber in the prehybridization solution (5 × SSC, 5 × Denhardt's solution, 250 μg ml–1 yeast tRNA, 4 mM EDTA, 0.8 μg ml–1 salmon sperm DNA, 0.3% Triton X-100, 50% deionized formamide) at 49C for 4 hr. Prehybridization was followed by hybridization in the solution having the same composition as the prehybridization mixture, except that salmon sperm DNA was replaced by the RNA antisense probe (200 ng ml–1). In control experiments we applied the same concentration of sense instead of anti-sense RNA probe at this step. After 16 hr of hybridization at 49C, samples were rinsed in SSC solutions of increasing stringency (twice for 15 min in 2 × SSC, 15 min in 0.2 × SSC, and twice for 15 min in 0.1 × SSC) at constant temperature (49C). The DIG-labeled probe was visualized by incubation with alkaline phosphatase-labeled anti-DIG antibody and subsequent alkaline phosphatase staining according to the protocol supplied by the producer (DIG Nucleic Acid Detection procedure; Boehringer-Roche). The concentration of anti-DIG antibody was 150 mU ml–1.
The described protocol proved successful for all in situ procedures in which RNA probes (AChE 1, AChE 2, and BuChE) were used for hybridization. For the oligonucleotide probes however, we failed to obtain any localization, although we tested several additional prehybridization and hybridization variants. We tested different temperatures and times of prehybridization and hybridization, two different concentrations of the probes, various times of incubation with anti-digoxigenin antibodies, and various times of alkaline phosphatase staining. Because none of these variants provided satisfactory localization, we do not describe them here in detail.
Nuclear Staining
ISH of AChE mRNA was combined with fluorescent staining of cell nuclei. By labeling cell nuclei we were able to determine the number of cells in the spinal cord sections and to identify cell types on the basis of the morphological characteristics of their nuclei. Glial cells and neurons could be easily recognized and differentiated on the basis of this criterion. Nuclei were stained after the completed ISH procedure with 1 μg/ml Hoechst 33342 (Molecular Probes; Eugene, OR) in PBS for 5 min at room temperature. Sections were then washed five times with PBS, coverslipped with 70% glycerol, and examined under the microscope.
Determination of AChE mRNA and BuChE mRNA in the Rat Thoracic Spinal Cord by Competitive RT-PCR
Total RNA was isolated with the RNeasy Mini Kit (Qiagen; Valencia, CA) from tissue samples of approximately 20 mg weight. Pieces of thoracic spinal cord were disrupted by Ultra Turrax T25 (Janke & Kunkel) in lytic buffer containing guanidine isothiocyanate and β-mercaptoethanol and were homogenized with a QIAshredder homogenizer. Total RNA was purified from homogenate by affinity chromatography on a silica gel-based membrane supplied with the kit. RNA was eluted with 30 μl DEPC-treated water. Yield was determined by A260 readings. Using a first-strand cDNA synthesis kit (Reverse Transcription System; Promega, Madison, WI), mRNA was reverse transcribed by avian mieloblastosis virus-reverse transcriptase. Reverse transcription was primed with oligo-(dT) in a volume of 40–160 μl.
Oligonucleotide primers for the PCR experiments were purchased from Gibco (Grand Island, NY). Primer pairs were selected according to the melting temperature, GC content, and predicted product size. For AChE mRNA we used primers with the following rat sequences: forward 5′-GATCCCTCACTGAACTACACCGTGG-3′ (starts at nt 1471); reverse 5′-GGTTCTTCCAGTGCACCATGTAGGAG-3′ (starts at nt 1801). For BuChE mRNA we used primers with the following rat sequences: forward 5′-AGAATGGATGGGAGTAATGCATGG-3′ (starts at nt 1446); reverse 5′-GATGGAATCCTGCCTTCCAC
The levels of AChE mRNA and BuChE mRNA in the rat spinal cord were determined by competitive PCR, adding known amounts of internal standards to the PCR reaction mixture. Internal standards were DNA fragments that, after PCR amplification, had approximately the same length (496 bp for AChE and 495 bp for BuChE) as the PCR products amplified from cDNAs that were reverse-transcribed from AChE mRNAs and BuChE mRNAs, respectively. Internal standards competed with these cDNAs for the primers added to the PCR mixture because they had the same primer-binding sequences attached at their ends. By adding internal standard to the PCR reaction mixture at different but known concentrations, we were able to determine the equivalent point. This was the point at which electrophoretic bands of the PCR products corresponding to the respective AChE and BuChE cDNAs had the same intensities as the PCR products of the internal standards amplified in the same PCR reaction. At this point, the concentration of internal standard added to the PCR mixture equaled the concentration of AChE cDNA or BuChE cDNA in the mixture.
The final volume (25 μl) of PCR reaction mixture contained 0.1–3 μl of cDNA, 0.5 U of HotStarTaq DNA Polymerase (Qiagen), 200 μM dNTPs, 10 pmol of primers, and 1 × Q-Solution (Qiagen) in PCR buffer (Qiagen). After 15 min of DNA polymerase activation at 95C (MJ Research thermocycler), 35 cycles of amplification were performed with a final 10-min extension at 72C. Each PCR cycle consisted of denaturation at 94C for 1 min, annealing at 60 or 61C for 1 min and extension at 72C for 1 min. After electrophoresis in a 1% agarose gel, PCR products were examined for their sizes using a 100-bp DNA ladder marker (Promega). Gels were scanned (Hewlett Packard ScanJet 6200 C) and the relative optical densities of the bands were determined by ScionImage software. Equivalent point was determined from the relative densities with the help of Microsoft Excell.
Results
Validation of the ISH Procedure
By varying the prehybridization and hybridization conditions, we found that the acetylation step in the prehybridization is essential for elimination of nonspecific staining (compare Figures 1A and 1D), regardless of the probe used for hybridization. However, the correct, generally established pattern of AChE mRNA-positive cells in the striatum, which was used as a standard in our testings of experimental conditions (Figure 1), was obtained only with the RNA probes. In spite of several trials in which we tested a variety of conditions, we failed to localize AChE mRNA in this CNS region with digoxigenin-labeled oligonucleotides. Staining (not shown) that was occasionally observed under these conditions did not match the cellular distribution visualized by nuclear co-staining and was therefore artifactual. Failure to localize AChE mRNA with the nucleotide probe was not expected because Hammond et al. (1994), using oligonucleotides with the same sequence (antisense nts 1090–1152), reported correct AChE mRNA localization that was in accord with the reports of others (Landwehrmeyer et al. 1993; Bernard et al. 1995) as well as with our localization obtained with the RNA probe (Figure 1). The failure could be accounted for only by the labeling procedures because this was the part in which our procedure differed from that of Hammond et al. (1994). Instead of 3′ end-labeling of the probe with [33P]-dATP, as in the original study, we attached at the same 3′position a digoxigenin-tagged nucleotide tail. This tail obviously impaired the capacity of oligonucleotides to bind to their target mRNAs.
Localization of AChE mRNA in Rat Spinal Cord
Two probes were used for the AChE mRNA localization. The first one (AChE 1) was from the second exon and was therefore targeted to all AChE mRNA splice variants in the spinal cord. The second probe (AChE 2) spanned exons 4 and 6 and was therefore directed only to the mRNAs encoding the tailed (T) AChE form. Localization of AChE mRNA with the two probes was compared and analyzed in the ventral horns (roughly corresponding to areas VIII and IX, Rexed's classification), where we found most prominently stained AChE mRNA-positive cells identified in our analyses as motor neurons (see Figure 3A).
On average, we found 18 AChE mRNA-positive cells per ventral horn in one 25-μm spinal cord cross-section, which was about 1.25% of the total number of cells in the area. The general pattern of distribution of AChE mRNA positive cells and their types did not differ with regard to the AChE probe used for localization (Figure 2). This is consistent with the observation that the T splice variant is the principal AchE mRNA form in the adult rat CNS (Seidman et al. 1995). The intensity of staining of AChE mRNA-positive cells as well as their number appeared even higher with AChE 2 than with AChE 1 probe (compare Figures 2A and 2B, and Figures 2G and 2H), although the AChE 2 probe was directed only toward a subpopulation of the transcripts targeted by the AChE 1 probe. Staining of the spinal cord section with the AChE 1 probe resulted in almost exclusive staining of a limited population of cells in the ventral horns. Because these cells also exhibited the highest staining intensity with the AChE 2 probe (Figure 3A), we concluded that they had the highest AChE mRNA levels. On the basis of their size and nuclear morphology, we identified these cells as neurons (Figures 2C and 2D; Figures 5A, 5C, and 5E). Glial cells, identified by small compact nuclei (Figures 2C and 2D; Figures 5A, 5C, and 5E) were AChE mRNA-negative in our experiments. AChE mRNA-positive cells were also cholinergic because the types and number of cells that were AChE mRNA-positive correlated well with the ChAT-positive cells (not shown).
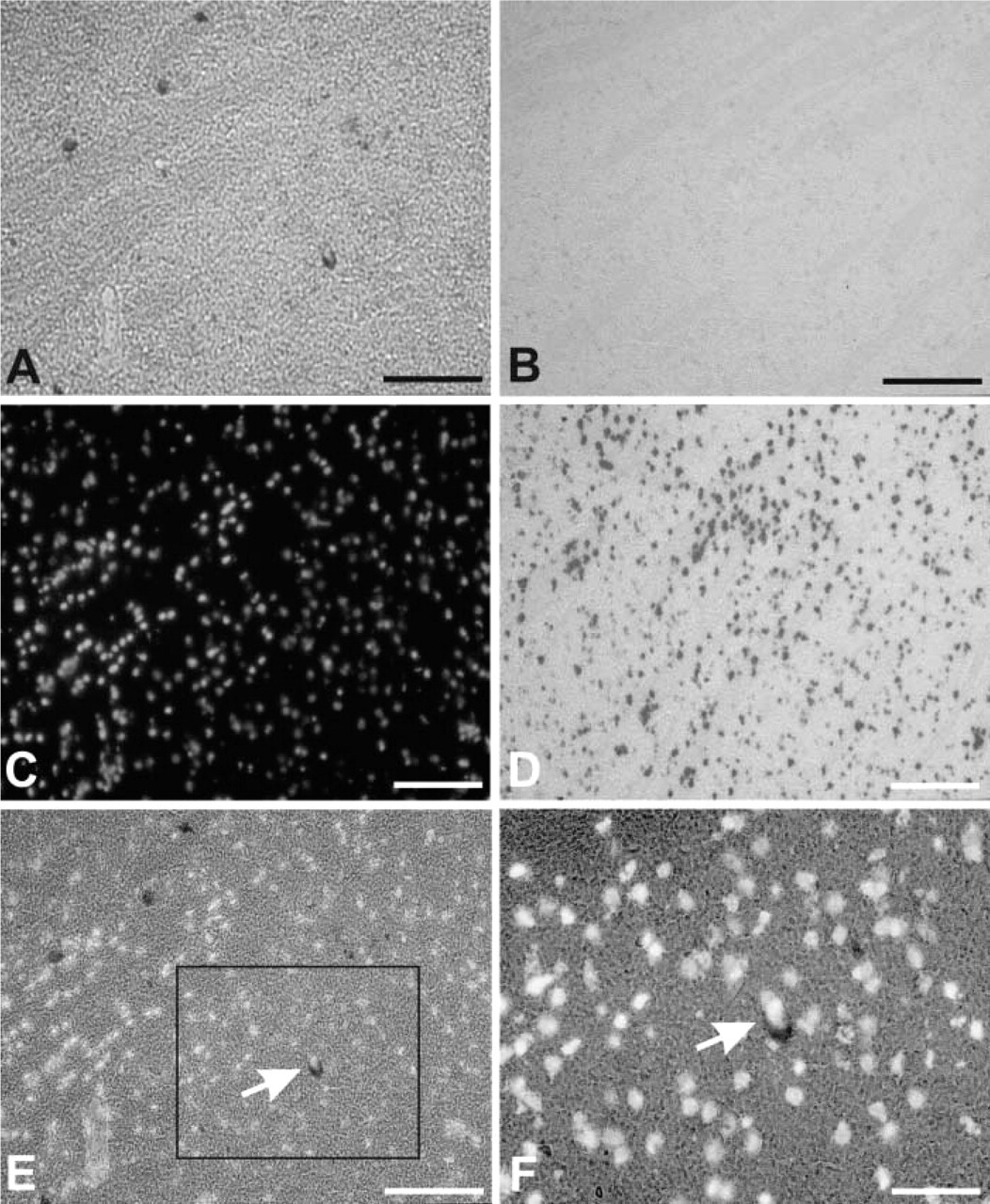
ISH of AChE mRNA in the rat striatum. (
With probe AChE 2 we found AChE mRNA-positive cells also in the lateral horn, where the perikarya of cholinergic preganglionic sympathetic neurons are located. AChE mRNA-positive cells were also found in the dorsal horns, but their staining was weaker than the staining of neurons in the ventral horns (Figure 3A).
Comparison of AChE mRNA and BuChE mRNA Localization in Rat Spinal Cord
ISH with specific RNA probes revealed the distribution of the cells expressing AChE mRNA (Figure 3A) and BuChE mRNA (Figure 3B) in the cross-sections of rat thoracic spinal cord. Combining this technique with fluorescent nuclear staining, we were also able first, to identify the cells expressing these transcripts as to whether they belonged to neurons or, to glia (Figures 5A–5F; see also Figures 2C and 2D) and, second, to determine their percentages (Figures 5G and 5H). Expression of both messages in the thoracic spinal cord was additionally demonstrated by RT-PCR. By using competitive approach in the RT-PCR experiments, we were also able to compare quantitatively the levels of AChE mRNA and BuChE mRNA in this part of the CNS (Figure 4).
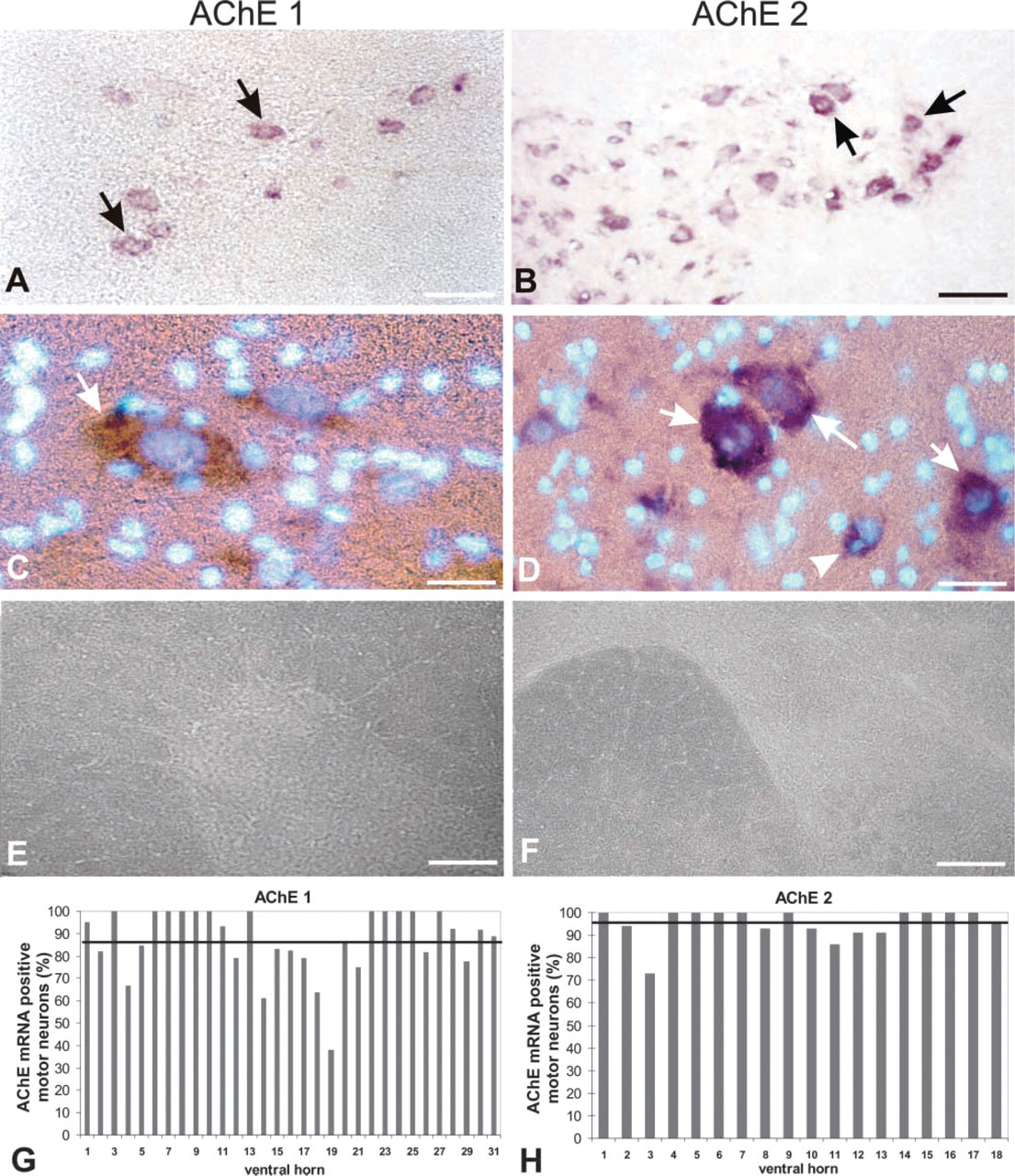
AChE mRNA localization in the ventral horns of cross-sections of rat thoracic spinal cord by probes targeted either to all AChE mRNA species (AChE 1 probe) or specifically to the tailed (T) AChE mRNA (AChE 2 probe). (
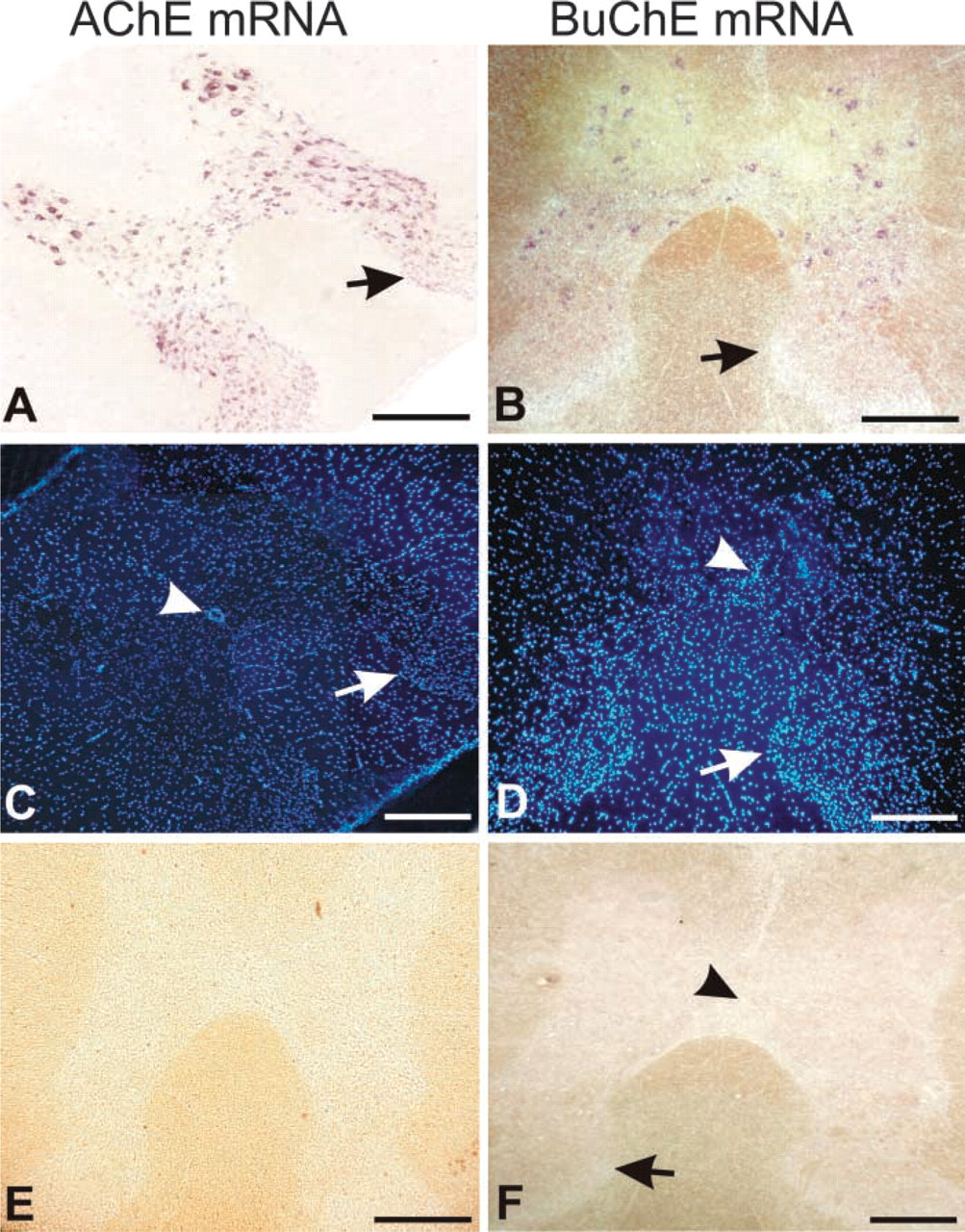
Localization of AChE mRNA and BuChE mRNA in cross-sections of the thoracic spinal cord. (
The number of BuChE mRNA-positive cells was lower than the number of AChE mRNA-positive cells. The difference appears clear at low magnification (compare Figures 3A and 3B). However, quantitative analyses (determination of mean percentages of positive cells ± SD; n = number of ventral horns examined) carried out on serial sections at the higher magnification (Figure 5) revealed that the percentage of BuChE mRNA-positive cells (85 ± 8%; n = 10; Figure 5H) was almost equal to the percentage of AChE mRNA-positive cells determined with the AChE 1 probe (87 ± 15%; n=31) (Figure 2G) and was only 12% lower than the percentage obtained by the AChE 2 probe (97 ± 7%; n=18; Figure 2H).
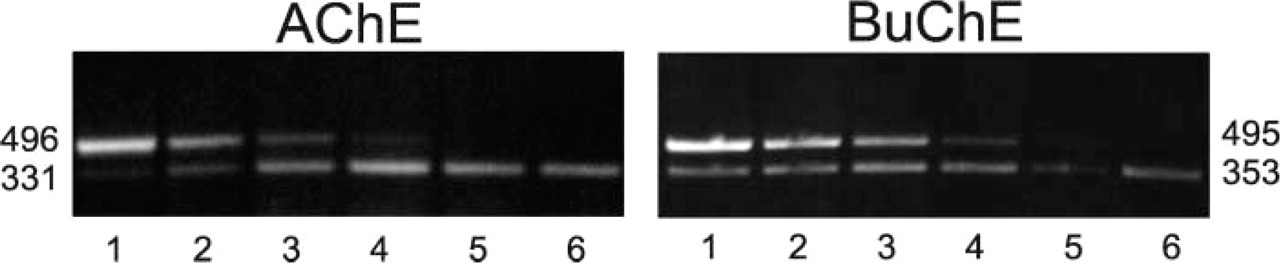
PCR products detected in the typical competitive RT-PCR determination of AChE mRNA and BuChE mRNA in rat thoracic spinal cord. Six lanes in each gel correspond to six PCR reactions run in parallel. AChE cDNA and BuChE cDNA, reverse transcribed from their corresponding mRNAs, were added to PCR reactants and the buffer and then divided into six aliquots. The internal standard DNAs, competing for the same primers, were added to the PCR reaction mixtures at concentrations falling from the highest in Lane 1 to the lowest in Lane 6. PCR products of AChE and BuChE cDNAs (lower bands) and their corresponding internal standards (upper bands) had the expected nucleotide lengths at left (AChE) and a right (BuChE) of the corresponding gels.
The general distribution pattern of mRNA-positive cells in the spinal cord did not differ between AChE and BuChE (Figure 3). Nonuniformity of staining, ranging from heavy to barely visible, was found in both AChE mRNA- and BuChE mRNA-positive cells (Figure 5). As in the case of AChE mRNA, we found by far the highest level of BuChE mRNA staining in α-motor neurons (Figures 5B, 5D, and 5F). A small population of neurons (between 3 and 15%) was negative for the transcripts of both AChE and BuChE. Glial cells were practically negative for BuChE mRNA (Figure 5) and AChE mRNA (Figures 2 and 5), although on some rare occasions we observed weak BuChE mRNA staining associated with the nuclei characteristic for glial cells (Figure 5).
By RT-PCR we also found higher levels of AChE mRNA in comparison to BuChE mRNA in the rat thoracic spinal cord. However, although the ratios of AChE mRNA to BuChE mRNA levels, as determined by this technique in whole thoracic spinal cords, were highly variable (in three determinations we found sevenfold, 15-fold, and 16-fold more AChE mRNA than BuChE mRNA), they were in all determinations much higher than the ratios of AChE mRNA, to-BuChE mRNA-positive cells determined by ISH (see above). Taken together, these results suggest that the cells expressing AChE mRNA in the rat thoracic spinal cord also express BuChE mRNA but that the levels of AChE mRNA are several-fold higher than the BuChE mRNA levels in these cells.
Discussion
Technical Considerations
This is the first study using mRNA localization to compare the distribution of cells expressing AChE and BuChE in a region of the mammalian CNS. Nonradioactive digoxigenin-based ISH, introduced for this purpose, proved suitable for the localization of AChE mRNA and BuChE mRNA in the rat spinal cord and might also prove useful for similar studies in other CNS regions. Our protocol is significantly shorter in comparison to radioactive techniques: it takes only 2 days from the isolation of tissue to the microscopy which is considerably less than the 6 to 10 days (Hammond et al. 1994; Bernard et al. 1995) or even 4 weeks (Landwehrmeyer et al. 1993) reported for radioactive ISH of AChE mRNA. Personnel are not exposed to radiation and, finally, nonradioactive labeling is more suitable for the experimental planning. Unlike radioactively labeled probes, which can be used only within a limited time span determined by the isotope half-life, nonradioactive probes can be used for a period of more than a year. A drawback of the technique is that it is sometimes unpredictable. For no obvious reason we sometimes obtained no staining with the antisense probes or unspecific staining in the control experiments with sense probes. Our interpretation is that the complexity of the technique, which includes several steps and several parameters that are not easy to control, in itself introduces a certain degree of variability. These problems can be overcome by including in further analyses only those ISH experiments in which we observed specific perinuclear staining of the cells with the antisense probe and no staining with the control sense probe. All experiments not fulfilling these criteria were discarded.
Only RNA probes worked in our experiments. We were unable to achieve specific localization with the oligonucleotides labeled at the 3′ location with digoxigenin-tagged nucleotides. We speculate that altered biochemical and biophysical properties of oligonucleotides due to attachment of the long lypophilic tail (see Materials and Methods) as a result of labeling caused their failure to bind to their target mRNAs.
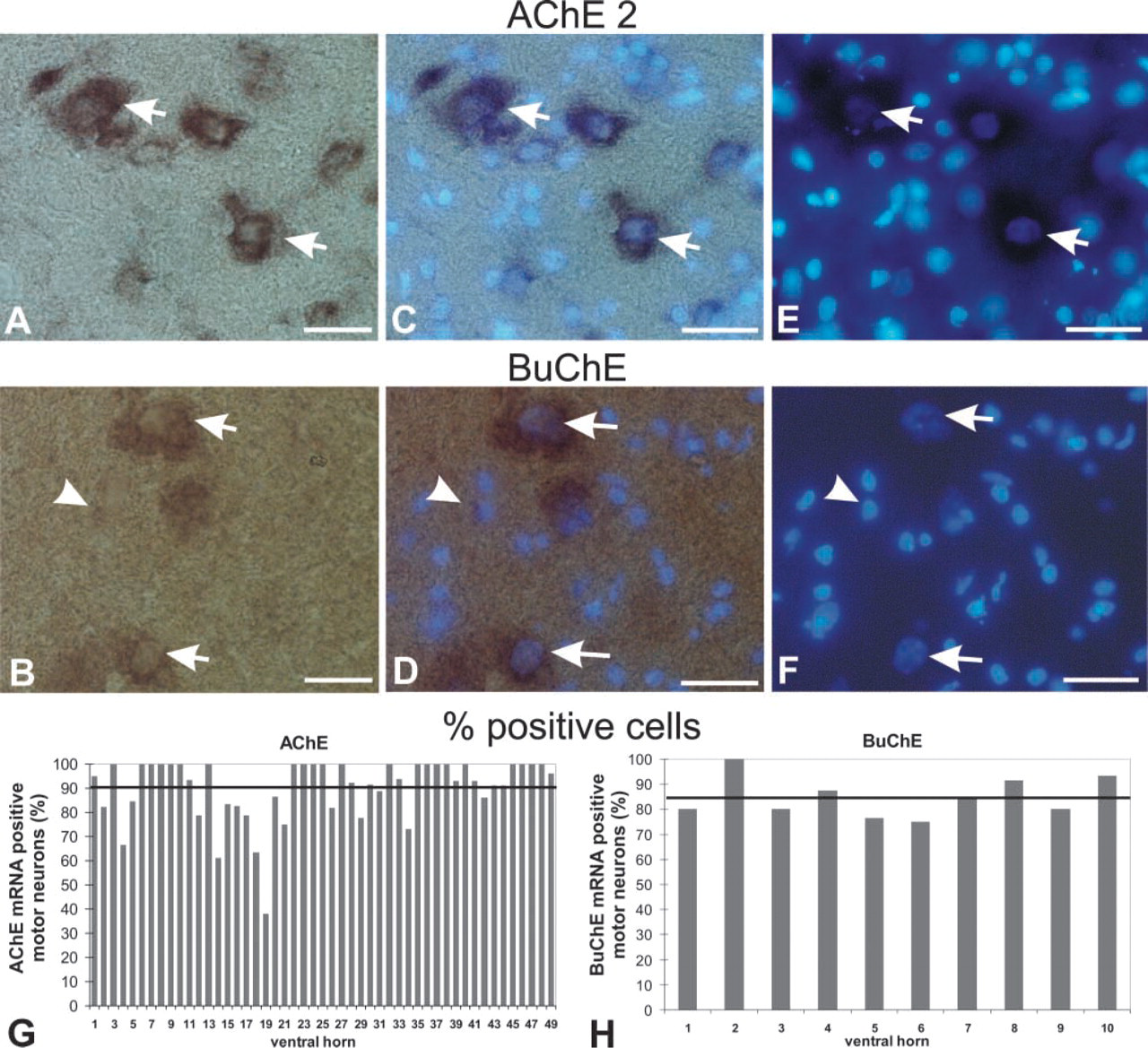
Identification of cells expressing (
With respect to acetylation, we found dramatically reduced nonspecific background staining after the introduction of this reaction into the prehybridization step. Acetylation is frequently used in the ISH procedures to reduce nonspecific staining. It has been proposed that this reaction coats the section with a negative charge that may repel negatively charged nucleic acid probes (Wisden et al. 1991). However, this proposal does not explain why acetylation is so effective when ISH probes are labeled with digoxigenin. One possibility is that acetic anhydride, which is a powerful agent in the presence of negatively charged moieties, actually removes negatively charged elements in the tissue, which therefore minimizes nonspecific reaction between negative charges on the tissue and digoxigenin.
On the basis of nuclear morphology, as revealed by fluorescent staining of the cell nuclei by Hoechst 33342, we identified on average 18 AChE mRNA-positive motor neurons in both ventral horns per 25-μm section. Larger nuclei were indicative for the α-motor neurons, while the smaller ones most probably belonged to γ-motor neurons. Because there was also a small percentage (3–15%) of neurons that did not stain for AChE mRNA, our identification was in fairly good agreement with the number of motor neurons in rat spinal cord (22 ± 11) reported previously (Delfs et al. 1989). This simple identification, which can easily be combined with ISH, should therefore prove suitable in other studies in the CNS to differentiate among neuronal and glial cells with regard to their expression of specific mRNAs.
Localization of AChE mRNA and BuChE mRNA in the Spinal Cord
The essential finding of this study is the identity of cells expressing AChE and BuChE mRNAs. However, from our RT-PCR determinations we estimate that the levels of AChE mRNA are several times higher than the levels of BuChE mRNA in these cells. Similar percentages of cells positive for AChE mRNA and BuChE mRNA therefore do not reflect the quantitative levels of these mRNAs, most likely because the intensity of ISH staining depends not only on the mRNA level in the cell but also on the staining reaction in which alkaline phosphatase, bound to anti-digoxigenin antibodies, produces a colored product. Only neurons were positive for both messages in the adult rat thoracic spinal cord. This is in good agreement with the report of Andres et al. (1997), who also found AChE mRNA expression only in neurons. Identifying neuronal subtypes on the basis of their morphology we found the highest staining intensity for both transcripts in α-motor neurons, although smaller ventral horn neurons, most likely γ-motor neurons, interneurons, and the preganglionic autonomic neurons located in the lateral horn also expressed both messages. The percentage of AChE mRNA cells was slightly higher with AChE 2 than with the AChE 1 probe, although it was directed only to the AChE mRNAs encoding T-isoforms and not to the total AChE mRNA population like the AChE 1 probe. Although this observation suggests a higher sensitivity of the AChE 2 probe, this difference might not be related to the differences in probes but rather to the intrinsic and expected variations in sensitivity among individual ISH experiments. The distribution patterns of AChE mRNA and BuChE mRNA positive cells corresponded well to the distribution of cholinergic ChAT-positive cells described previously by Sofroniew et al. (1985) and confirmed in our experiments. Many previous studies have already shown that cholinergic cells are the main producers of AChE (reviewed in Silver 1974; Chatonnet and Lockridge 1989; Taylor and Radic 1994; Darvesh et al. 2003). Here we demonstrate that cholinergic cells also produce BuChE.
We observed practically no staining in glial cells. Only occasionally did we observe weak BuChE mRNA staining associated with the nuclei characteristic for glial cells. This observation suggests that BuChE mRNA or even AChE mRNA is present in glial cells, although at levels not high enough to reach the sensitivity limit of detection. Nevertheless, considering the widely recognized high sensitivity of ISH, these transcripts, even if they exist, are expressed at very low levels.
Discussing the absence of transcripts encoding AChE and BuChE in the glial cells, we must also bear in mind that low levels of mRNA are not necessarily indicative of low levels of the mature protein, especially if mRNA turnover substantially exceeds protein turnover, as demonstrated for AChE in the skeletal muscle (Fuentes and Taylor 1993). AChE mRNA could not be detected in more than 50% of individual quail end-plates by RT-PCR, although AChE levels were high in every single end-plate (Jasmin et al. 1993). To explain this discrepancy the authors proposed that transcription of AChE mRNA occurs intermittently rather than constitutively. We found a similar relationship between AChE mRNA and mature enzyme in in vitro innervated human muscle (Grubic et al. 1995). Our observation that between 3–15% of neurons were negative for AChE mRNA and BuChE mRNA, respectively, and that the mRNA-positive neurons exhibit high nonuniformity of staining, suggests that the same, i.e., an intermittent pattern of AChE mRNA expression, might also take place in the CNS. Therefore, it cannot be excluded that occasionally, and at much lower frequency than in neurons, BuChE mRNA and perhaps also AChE mRNA are expressed in glia.
Demonstration of the mRNAs encoding AChE and BuChE in the motor neurons is in agreement with the report of Flumerfelt and Lewis (1975), who detected both AChE and BuChE activity in motor neurons in rat hypoglossal nucleus. In general, however, there is a great variety in the localizations reported for both enzymes in various parts of the CNS. Thus, in normal aged and Alzheimer's disease-affected human cerebral cortices, Wright et al. (1993) demonstrated AChE and BuChE activities in the astrocytes and oligodendrocytes. Activity ratios of the two enzymes differed among these cells according to the cortical region. AChE activity in neurons and BuChE activity in the non-neuronal elements, such as Schwann cells and neuropil, has been demonstrated in the rat dorsal root ganglia (Koenigsberger et al. 1998) and in the various brain areas of wild-type and AChE-nullizygote mice (Mesulam et al. 2002). Differences in AChE and BuChE distributions were also found in human amygdala and hippocampus, but this time the localization was reversed. BuChE was located primarily in neurons and AChE in neuropil (Darvesh et al. 1998). All these variabilities suggest species and regional specificity of the AChE and BuChE localization, which do not allow generalization of our findings to the entire CNS.
The high levels of AChE mRNA and BuChE mRNA in the α-motor neurons suggest that these cells need more AChE and BuChE than other cells in the spinal cord. Because α-motor neurons participate in the formation of the neuromuscular junction (NMJ) and its maintenance, these additional needs might be due to the neuronal contribution of these two enzymes to the NMJ. Such a contribution has been proposed before (Anglister 1991) and was also demonstrated in our laboratory (Jevsek et al. 2002). Membrane-associated BuChE that persists in the NMJs in ColQ-deficient mice (Feng et al. 1999) is therefore more likely to be of neuronal than of glial origin. Co-expression of both cholinesterases in the same cell types is consistent with the AChE back-up role proposed for BuChE by Xie et al. (2000).
Footnotes
Acknowledgements
Supported by a grant to Zoran Grubic from the Ministry of Education, Science, and Sport of Slovenia.
The technical assistance of Ms Zvonka Frelih is gratefully acknowledged.