
Abstract
Perlecan is a major heparan sulfate proteoglycan (HSPG) of basement membranes (BMs) and connective tissues. The core protein of perlecan is divided into five domains based on sequence homology to other known proteins. Commonly, the N-terminal domain I of mammalian perlecan is substituted with three HS chains that can bind a number of matrix molecules, cytokines, and growth factors. Perlecan is essential for metazoan life, as shown by genetic manipulations of nematodes, insects, and mice. There are also known human mutations that can be lethal. In vertebrates, new functions of perlecan emerged with the acquisition of a closed vascular system and skeletal connective tissues. Many of perlecan's functions may be related to the binding and presentation of growth factors to high-affinity tyrosine kinase (TK) receptors. Data are accumulating, as discussed here, that similar growth factor-mediated processes may have unwanted promoting effects on tumor cell proliferation and tumor angiogenesis. Understanding of these attributes at the molecular level may offer opportunities for therapeutic intervention.
Keywords
B
Structure of Perlecan
Perlecan was first isolated from a mouse Engelbreth-Holm-Swarm (EHS) tumor as a large low buoyant density HSPG (Hassell et al. 1980). It is widely distributed in the BMs of skin (Figure 1), breast, heart, pituitary gland, thymus, prostate, colon, lung, kidney, ear, and placenta. All vascular BMs in these tissues and organs contain perlecan. Perlecan is also present in some connective tissues that lack classical BMs, such as cartilage matrix and sinusoids of liver, spleen and lymph nodes (Murdoch et al. 1994; Couchman et al. 1995). A number of mammalian cell types can synthesize perlecan, such as epithelial (Mohan and Spiro 1991), epidermal (Tapanadechopone et al. 2001), and endothelial cells (Knox et al. 2002), smooth muscle cells (Weiser et al. 1996), fibroblasts (Tufvesson and Westergren-Thorsson 2000), and chondrocytes (SundarRaj et al. 1995). The various cellular sources of perlecan contribute to the ubiquitous distribution of perlecan in BMs and connective tissues.
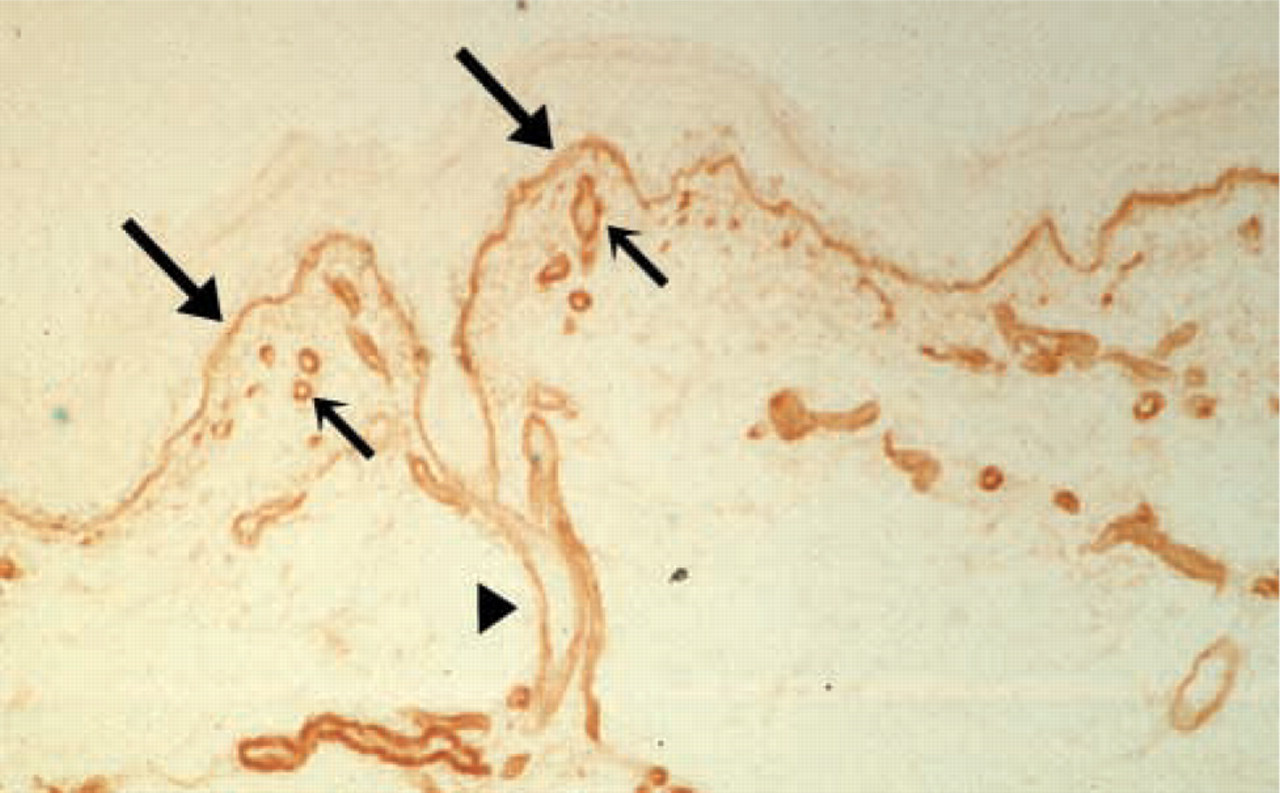
Human skin section stained with perlecan antibody. The distribution of perlecan in the dermal-epidermal junction (large arrows), capillaries (small arrows), and hair follicle (arrowhead) are indicated.
Perlecan Gene
The complete sequences of mouse (Noonan et al. 1991) and human (Kallunki and Tryggvason 1992; Murdoch et al. 1992) perlecan were reported over a decade ago. Mammalian perlecan, encoded by the HSPG2 gene, is a single-copy gene located on the short arm of human chromosome 1 at 1p36.1-p35 (Dodge et al. 1991; Kallunki et al. 1991) and on a syntenic region of mouse chromosome 4 (Chakravarti et al. 1991). Human perlecan gene consists of 94 exons and spans more than 120 kb of genomic DNA (Iozzo and Murdoch 1996). The intron-exon junctions in the various modules of human perlecan are remarkably conserved (Dodge et al. 1991; Kallunki et al. 1991), suggesting that the perlecan gene might have evolved from an ancestral gene by gene duplication or exon shuffling. The core protein of mouse perlecan is encoded by a ∼12-kb mRNA with a deduced MW of 396 kD (Noonan et al. 1991). Human perlecan core protein is encoded by a 14.35-kb mRNA with a deduced MW of 467 kD (Kallunki and Tryggvason 1992; Murdoch et al. 1992).
Perlecan homologues in the invertebrates C. elegans (Rogalski et al. 1993; Mullen et al. 1999) and Drosophila (Adams et al. 2000; Friedrich et al. 2000; Voigt et al. 2002) have also been identified. Nematode perlecan, encoded by the unc-52 gene, spans more than 20 kb of genomic DNA on chromosome II and consists of 37 exons. The longest potential open reading frame of the unc-52 gene encodes a 3375-amino-acid protein with a MW of ∼370 kD (Mullen et al. 1999). Drosophila perlecan is encoded by the trol (terribly reduced optic lobes) gene, which has also been known in FlyBase as pcan and CG7981. Located on chromosome X or 1, trol has 34378 bp of genomic sequence and an mRNA of 12.6 kb (Adams et al. 2000; Voigt et al. 2002). A 450-kD protein has been detected in Drosophila cell lines Kc1, Er1, and embryo extract by using an antibody against Drosophila perlecan domain V (Friedrich et al. 2000).
Structure of Perlecan Core Protein
The core protein of mammalian perlecan is divided into five domains based on sequence homology to other known proteins (Hassell et al. 1980; Noonan et al. 1991; Kallunki and Tryggvason 1992; Murdoch et al. 1992) (Figure 2). The N-terminal domain I is unique to perlecan; it contains an SEA (
Domain II of perlecan has homology to the class A low-density lipoprotein (LDL) receptor (LA). It consists of four LA modules and one immunoglobulin (Ig)-like repeat. Domain III is homologous to part of laminin α-chains, containing three laminin domain IV-like modules (L4) and eight laminin epidermal growth factor (EGF)-like repeats (LE). Domain IV of perlecan is the largest, containing a long series of Ig-like repeats, similar to those of the cell adhesion molecule N-CAM. The number of Ig repeats varies among species. Human perlecan has 21 repeats, while mouse perlecan has 14 repeats, lacking seven repeats corresponding to the midway through the fifth to the twelfth repeats of human perlecan. The C-terminal domain V has similarity to the globular domain of laminin α-chain and agrin. It contains three laminin G domain-like modules (LG) and four EGF-like repeats (EG).
Invertebrate perlecans have a similar modular structure. Domain I of nematode perlecan/UNC-52 (Rogalski et al. 1993; Mullen et al. 1999) is a short, 28-amino-acid region rich in aspartic acid residues, but no potential glycanation sites are present. Domain II contains three LA repeats flanked by two Ig-like repeats, domain III has two L4 modules and seven LE repeats, and domain IV contains up to 15 Ig-like repeats. Domain V consists of three LG modules, three EG modules, and one threonine-rich region that is not present in mammalian perlecan (Figure 2).
Drosophila perlecan (Adams et al. 2000; Friedrich et al. 2000; Voigt et al. 2002) apparently lacks a domain I equivalent, but domain II contains at least 22 LA modules and two Ig-like repeats. Domain III has three L4 modules and seven LE repeats, and domain IV contains twelve Ig-like repeats. The C-terminal domain V contains three LG modules, three EG repeats, and a 30-residue sequence containing seven proline but no cysteine residues (Figure 2). Domains II-V of perlecan are therefore well conserved but mammalian domain I has no invertebrate equivalent.
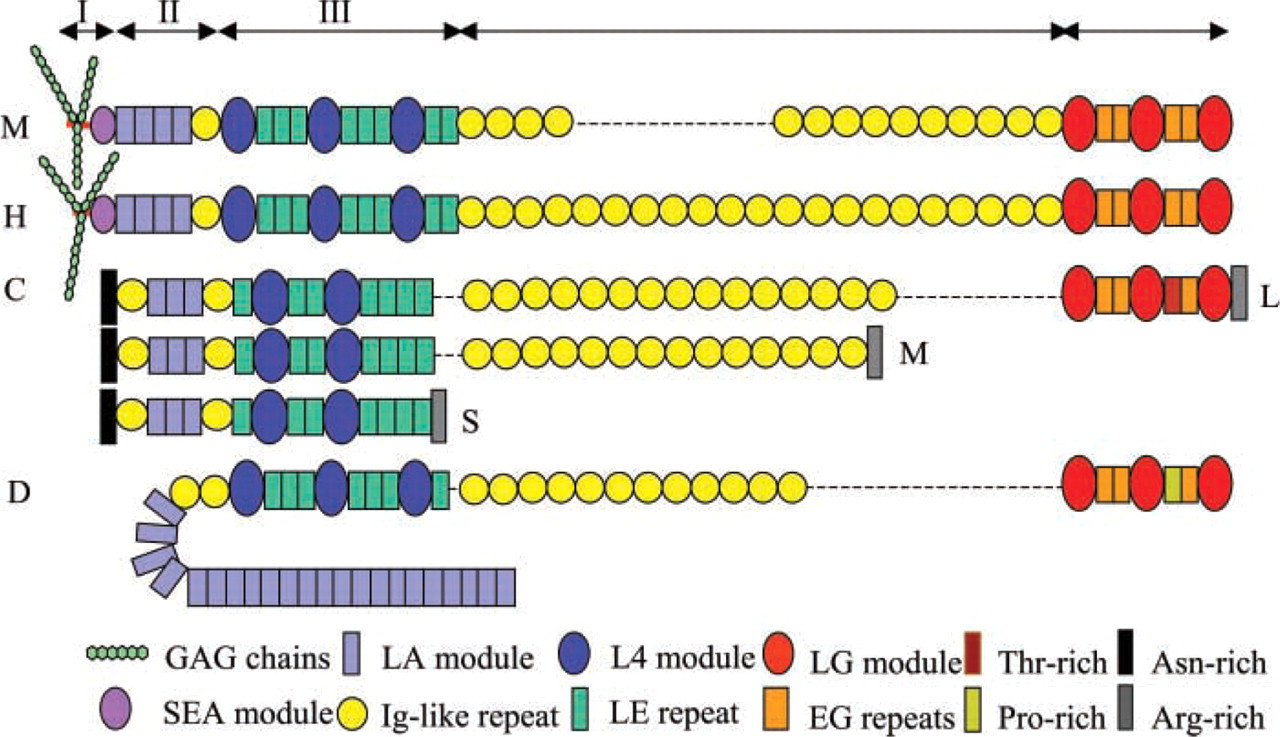
Schematic structures of perlecan from mouse (M), human (H), C. elegans (C), and Drosophila (D). Dashed lines, no corresponding sequence.
Alternative Splicing of Perlecan
Alternatively spliced perlecan variants have been found in mouse and nematode. Noonan and Hassell (1993) found three additional Ig-like repeats while screening the EHS tumor cDNA libraries, and these aligned well with human Ig-like repeats 8–10. Larger mRNA transcripts have been identified in mouse EHS tumor tissue and melanoma M2 cells, indicating that alternative splicing occurs in domain IV. Complex patterns of alternative splicing have been observed in nematode perlecan/UNC-52. Mullen et al. (1999) reported that perlecan/UNC-52 isoforms can be divided into three major groups. The long isoforms contain all five domains, the medium isoforms contain the first four domains, and the short isoforms contain the first three domains (Figure 2). In addition, alternative splicing of exons 6, 16, 17, 18, 21, and 22 results in more diversity within domains III and IV (Rogalski et al. 1993,1995). The alternative splicing of exons 16, 17, and 18 is regulated by the mec-8 (Lundquist et al. 1996; Spike et al. 2002) and smu-1 genes (Spike et al. 2001). The mec-8 gene encodes a nuclear protein that promotes the splicing of exon 15 directly to 19 or exon 16 directly to 19, resulting in the accumulation of a specific subset of alternatively spliced perlecan/ unc-52 transcripts (Lundquist et al. 1996; Spike et al. 2002). A presumptive null mutation in smu-1 suppresses nonsense mutations in exon 17 but not in exon 18, indicating that SMU-1 regulates the splicing of exon 17, which is skipped in the absence of functional SMU-1 (Spike et al. 2001).
Unlike mouse and nematode perlecan, human perlecan variants have not been observed despite extensive searching (Iozzo et al. 1994), and there are no data regarding potential alternative splicing of Drosophila perlecan.
Glycosaminoglycan Substitutions
Mammalian perlecan is predominantly substituted with HS chains, but on some occasions it may be substituted with CS, dermatan sulfate (DS), hybrid HS/CS, CS/DS chains, or secreted as GAG-free glycoprotein (Isemura et al. 1987; Iozzo and Hassell 1989; Kokenyesi and Silbert 1995; Couchman et al. 1996; Groffen et al. 1996; Dolan et al. 1997; Graham et al. 1999). While the major GAG attachment sites are located on domain I, domain V can be substituted with HS and/or CS chains as well (Brown et al. 1997; Friedrich et al. 1999; Tapanadechopone et al. 1999). Depending on the state of cell differentiation or tissue location, perlecan can possess GAG chains of various length and sulfation (Hassell et al. 1980; Mohan and Spiro 1991; Molist et al. 1998). Despite lacking domain I with its SGD peptide motifs, Drosophila perlecan may also be substituted with HS chains (Friedrich et al. 2000), although the sites are unknown. No information about the GAG substitution on perlecan/UNC-52 has been reported thus far.
Perlecan Interactions with Extracellular Matrix Components
Perlecan plays important structural role in BMs through its HS chains and core protein. The HS chains interact with a number of ECM molecules including laminin, type IV collagen, and fibronectin (Battaglia et al. 1992; Ettner et al. 1998). Domain IV can bind entactin/nidogen-1 and −2, fibulin-2, and fibronectin (Hopf et al. 2001), and domain V binds fibulin-2 and laminin-entactin/nidogen complex (Friedrich et al. 1999). The binding sites for perlecan on entactin/nidogen-1 have been mapped to the G2 and G3 domains (Reinhardt et al. 1993). It is suggested that BM is composed of two networks, one formed by laminin, and the other formed by type IV collagen. These two networks are connected by entactin/nidogen (Yurchenco and O'Rear 1994). Perlecan makes such supramolecular architecture more stable by interacting with laminin, type IV collagen, and entactin/nidogen. A decreased level of functional perlecan results in abnormal embryonic development (see below).
Perlecan Genetics
Embryogenesis studies of mice, C. elegans, and Drosophila revealed the timing of perlecan expression in BMs and connective tissues during development.
Genetics of Mammalian Perlecan
During murine embryogenesis, expression of perlecan is detected at embryonic day10.5 (E10.5) in tissues undergoing vasculogenesis, including heart, pericardium, and major blood vessels. Between E11 and E13, the strongest expression of perlecan is observed in developing cartilage, especially those undergoing endochondral ossification, in which levels remain elevated until adulthood. At later stages (E13-17.5), the expression of perlecan mRNA correlates with the onset of differentiation of various parenchymal tissues, including the developing kidneys, liver, spleen, lungs, and gastrointestinal tract (Handler et al. 1997). French et al. (1999) demonstrated that perlecan not only is a marker of chondrogenesis but also potentiates chondrogenic differentiation in vitro. When plated on perlecan matrix, multipotential murine fibroblast cells become rounded and aggregate into dense nodules (reminiscent of embryonic cartilaginous condensations) that stain intensely with Alcian blue and for type II collagen (chondrocyte marker), while plastic and other ECM components are unable to induce nodules. Heparinase rather than chondroitinase treatment of perlecan results in delayed and incomplete nodule formation. These data suggest that both perlecan core protein and HS chains are required for the chondrogenic activity in vitro (French et al. 1999). Recently, the chondrogenic activity of perlecan has been mapped to an HS-bearing domain I (French et al. 2002).
The crucial roles of perlecan in murine vasculogenesis and chondrogenesis are confirmed by two separate gene knockout analyse (Arikawa-Hirasawa et al. 1999; Costell et al. 1999). In perlecan-null mice, severe chondrodysplasia develops in embryos and BMs deteriorate in regions of increased mechanical stress, leading to pericardial leakage and exencephaly. The embryos die at E10-E12 or perinatally due to respiratory failure. Further evidence comes from two classes of human skeletal disorders. Dyssegmental dysplasia, Silver-handmaker type (DDSH) (Arikawa-Hirasawa et al. 2001a,b) and Schwartz-Jampel syndrome (SJS) (Nicole et al. 2000; Arikawa-Hirasawa et al. 2002a) are both linked to perlecan mutations. DDSH is a neonatal lethal autosomal recessive disorder, and approximately 20 cases of DDSH have been reported thus far (Arikawa-Hirasawa et al. 2001a). The embryos show similar skeletal abnormalities to perlecan-null mice. Analysis reveals that no perlecan is secreted into the ECM. Instead, it is degraded to smaller fragments intracellularly, indicating that DDSH is caused by a functional null mutation of perlecan (Arikawa-Hirasawa et al. 2001b). SJS is an autosomal recessive disorder characterized by various degrees of myotonia and chondrodysplasia. Analysis of fibroblasts and muscle cells from SJS patients showed that truncated perlecan, which lacks domain V or has 35- or 64-amino-acid deletions from the C-terminal of domain V, is secreted but in markedly reduced concentrations (Nicole et al. 2000; Arikawa-Hirasawa et al. 2002a). SJS patients can survive. Arikawa-Hirasawa et al. (2002b) reported that acetylcholinesterase is absent from the neuromuscular junctions of perlecan-null mice, indicating that perlecan can regulate the clearance of neurotransmitter by recruiting acetylcholinesterase through HS chains (Rossi and Rotundo 1996; Peng et al. 1999). These data may explain the myotonia suffered by SJS patients. The phenotypic differences between DDSH and SJS patients suggest that the level of functional perlecan in BMs and ECM is critical for musculoskeletal function.
Genetics of Nematode Perlecan/UNC-52
Unlike mammalian perlecan, which is distributed in all BMs, nematode perlecan/UNC-52 is not found in the BMs surrounding the intestine or lining the pseudocoelom but is specifically associated with muscle tissues (Mullen et al. 1999), which express perlecan/UNC-52 autonomously (Moerman et al. 1996). During embryonic development, perlecan/UNC-52 staining is first observed at the comma stage, located primarily at cell-cell contacts between muscle cells. From the comma to the 1.5-fold stage, perlecan spreads from cell-cell contacts to the basal surface of muscle cells, where the BM is located. At later stages, body wall muscle, anal muscle, and pharynx show positive staining (Mullen et al. 1999).
Mutation analysis shows that the medium perlecan/UNC-52 isoforms are sufficient enough for proper embryonic and larval development (Mullen et al. 1999). To date, more than 10 unc-52 mutations have been identified (Rogalski et al. 1995; Mullen et al. 1999). Null mutations lead to Pat (paralyzed, arrested elongation at twofold), a lethal phenotype. In the body wall muscle cells of mutant embryos, myosins form large aggregates instead of ordered bands, indicative of a failure in myofilament lattice assembly (Rogalski et al. 1993; Mullen et al. 1999). Viable mutations of unc-52 are clustered in a small region containing exons 16, 17, and 18. Each viable mutation affects only some but not all of the perlecan/UNC-52 isoforms (Rogalski et al. 1995). Viable mutants are able to move normally as larvae but adults are paralyzed (Gilchrist and Moerman 1992). Beginning at the third or fourth larval stage, the body wall muscle cells posterior to the pharynx gradually disrupt, correlating with progressive paralysis. The dense bodies in the affected muscle cells are fractured and the myofilaments lose attachment to the cell membrane (Waterston et al. 1980). Loss of function mutations in mec-8 enhances the mutant phenotypes of viable unc-52 mutations due to the inability of mutant mec-8 to skip exons 16, 17, and 18. The double mutants exhibit similar phenotypes to unc-52 null mutants (Lundquist and Herman 1994) and have severely reduced levels of functional perlecan/UNC-52 (Mullen et al. 1999). The null and viable mutation phenotypes suggest that perlecan/UNC-52 plays an essential role in myofilament assembly in body wall muscle during embryonic development.
Genetics of Drosophila Perlecan
In Drosophila, trol mRNA is first detected in the syncytial blastoderm during embryonic development (Friedrich et al. 2000). At stage 15, high expression is observed in the visceral mesoderm of the gut and in the cardiac cells. Perlecan protein is first detected at this stage, with particularly strong expression around the central nervous system (CNS), the visceral mesoderm, and the hindgut. Perlecan may regulate the timing of neuroblast proliferation in Drosophila. In trolsd mutants, quiescent optic lobe and thoracic neuroblasts fail to begin proliferation, resulting in a dramatic decrease in the number of dividing neuroblasts in the larval brain (Datta 1995). The trolsd mutation is lethal and the mutant has a phenotype of aberrant larval brain lobe morphology and small imaginal discs (Datta and Kankel 1992). Further studies showed that induced expression of cyclin E in trol mutants results in normal levels of dividing neuroblasts in the larval CNS (Caldwell and Datta 1998), suggesting that perlecan may regulate the reactivation of neuroblast proliferation by stimulating the G1/S-phase transition through upregulation of cyclin E expression. To date, about 140 mutant trol alleles have been recorded (Voigt et al. 2002). They affect the larval CNS, brain, ventral ganglion, and thoracic neuroblasts.
Perlecan therefore has multiple roles in each species examined. It appears that the acquisition of a closed vascular system and extensive skeletal connective tissue in vertebrates has led to further roles for this proteoglycan. The common determinants for many of these functions, however, may be the GAG chains and their ability to bind growth factors. This feature may also underpin the accumulating evidence that there are roles for perlecan in tumor growth, and tumor angiogenesis.
Perlecan and Tumor Angiogenesis
Perlecan Expression in Tumors
Expression of perlecan is enhanced in a number of tumor types, both in vivo and in vitro. In human salivary adenoid cystic carcinoma (ACC), which is histopathologically characterized by cribriform structures composed of pseudocytes, perlecan mRNA is highly expressed in cuboidal carcinoma cells forming small pseudocytes, whereas the signals are absent in flat carcinoma cells forming large pseudocytes or carcinoma cells attaching to the peripheral nerves, suggesting that perlecan might be required for the growth of ACC cells. When the perlecan level is low in the intercellular space of small pseudocytes, ACC cells synthesize perlecan and secrete it into the intercellular space that might maintain their own growth. However, no perlecan production may be necessary in large pseudocytes and peripheral nerves where abundant perlecan has already been deposited (Kimura et al. 2000). In human ameloblastomas, perlecan core protein is detected in the intercellular space of stellate reticulum-like cells, which are located in the center of tumor cell nests. Meanwhile, perlecan is located more intensely in the myxofibrous stroma, especially around tumor cell nests. Perlecan mRNA is detected in both tumor cells and stromal fibroblasts, and signals are most enhanced at the periphery of tumor cell nests, especially in the invading fronts of branching nests (Ida-Yonemochi et al. 2002). In human intrahepatic cholangiocarcinoma (ICC), and liver carcinoma metastatic from the colon, strong expression of perlecan core protein is detected in tumor stroma, whereas only small amounts of perlecan are detected in normal liver around the bile duct and the blood vessels in the portal area. At the mRNA level, signals for perlecan are found in both carcinoma cells and stromal myofibroblasts. Similar to human ameloblastomas, the mRNA signals in ICC are more intense in the carcinoma cells at the invading fronts (Sabit et al. 2001), indicating that perlecan is involved in the invasiveness of these tumor cells. In human metastatic melanomas, perlecan mRNA level is markedly increased compared to normal tissue. The over-expressed perlecan, which is deposited in the ECM, appears to be related to the invasiveness of the melanoma cells (Cohen et al. 1994). The upregulation of perlecan is also observed in vitro. The tumorigenic mouse epidermal cell line RT101 synthesized more perlecan at both protein and mRNA levels than its normal JB6 counterpart (Tapanadechopone et al. 2001). Marchetti et al. (1993) showed that the invasiveness of human melanoma cells was enhanced by purified perlecan, while downregulation of perlecan by antisense targeting reduced the proliferation and invasiveness of murine melanoma cells (Adatia et al. 1997). Systemic administration of rabbit anti-perlecan antibody 2 hr before the injection of mouse metastatic Lewis lung tumor cells into the heart ventricle of C57Bl/6 mouse inhibited kidney and liver colonization (Tovari et al. 1997). These results indicate that perlecan is positively correlated with tumor growth, invasion and metastasis.
However, the expression of perlecan core protein is undetectable in invasive human lung carcinomas (Nackaerts et al. 1997). Nerlich et al. (1997,1998) reported that high levels of perlecan, collagen type IV, and fibronectin mRNAs were detected in both tumor and stromal cells of human breast and laryngeal carcinomas, but very low levels of perlecan, collagen type IV, and fibronectin protein were present in the fragmented tumor BMs. This might be a consequence of enhanced proteolytic degradation of BM components or post-transcriptional inhibition of protein synthesis. The loss of perlecan immunoreactivity in the fragmented BMs of human breast cancer is contrary to observations of human breast cancer stroma, in which abundant deposits of perlecan were detected (Iozzo et al. 1994). The other controversial reports regarding the perlecan levels are related to human hepatocellular carcinoma (HCC). Sabit et al. (2001) showed that perlecan core protein was mostly localized in the fibrous tissue surrounding HCC nodules, but carcinoma cells and their stromal space were negative for perlecan immunostaining. However, Roskams et al. (1998) demonstrated that strong perlecan immunoreactivity was detected in the BMs and tumoral stromal mesenchymal cells. The tumor subtypes, tumor stages, the degree of tumor cell differentiation, and/or various histological positions in the tumors may result in the different distribution patterns of perlecan observed in human breast cancer and HCC. The low level or absence of perlecan core protein in tumors suggests that perlecan might have inhibitory effects on tumor growth. In human fibrosarcoma cells, antisense suppression of perlecan expression resulted in stimulation of tumor cell growth, both in vitro and in vivo, and increased invasion of ECM (Mathiak et al. 1997). In human Kaposi's sarcoma cells, reduction of perlecan by antisense cDNA reduced cell migration and proliferation in vitro but increased tumor growth in vivo (Marchisone et al. 2000). In these cases, lack of perlecan may favor the diffusion of heparin-binding growth factors, leading to enhanced tumor growth. Therefore, the function of perlecan in tumor invasion and metastasis is dependent on the cellular context.
Perlecan and Tumor Angiogenesis
Tumor growth is often accompanied by angiogenesis, which is the process of forming new blood vessels from pre-existing blood vessels. Without new blood vessels, tumors cannot expand beyond a few millimeters, the point at which the diffusion of oxygen, nutrients, and the disposal of waste products become limiting (Folkman 1971). Perlecan may not only affect tumor invasion and metastasis but also support tumor angiogenesis.
Perlecan by itself is able to stimulate angiogenesis in a rabbit ear model of in vivo angiogenesis (Aviezer et al. 1994). In human papillary and invasive ductal breast carcinomas, large deposits of perlecan core protein are found in the tumor stroma and blood vessel walls (Guelstein et al. 1993). Similarly, abundant perlecan core protein is detected in the vessel walls and endothelial cells of human primary liver tumors (Roskams et al. 1998). What is the origin of perlecan deposited around the tumor blood vessels? As described previously, both tumor and host stromal cells have the potential to synthesize perlecan (Ida-Yonemochi et al. 2002; Nerlich et al. 1997,1998; Sabit et al. 2001). Tumor cells may secrete perlecan to provide a scaffold for blood vessel formation during tumor expansion, and perlecan deposited by tumor cells may further affect tumor growth by binding and modulating the activities of angiogenic growth factors. On the other hand, host cells may synthesize perlecan as a defensive mechanism to limit the diffusion of angiogenic growth factors. Data from Iozzo's group (1994) gave a first clue about the origin of perlecan in tumor blood vessels. They injected human prostate carcinoma cells into mice, and found that perlecan deposited along the BMs of newly formed blood vessels in tumor xenografts is of human origin. Interestingly, gradient perlecan immunoreactivity is observed in the tumor vessels, with the most immunoreactive microvessels located at or around the growing edge, suggesting that tumor-derived perlecan can induce or favor the neovascularization of the tumor. Does host tissue also synthesize perlecan and deposit it into the tumor blood vessels? To address this issue, we injected RT101 tumor cells intradermally into immunocompromised rats and immunostained the tumor cryosections with species-specific monoclonal antibodies. Perlecan deposited around blood vessels was of both tumor and host origin. Additionally, tumor cell-derived perlecan was extensively distributed throughout the tumor matrix (Figure 3). Several questions emerge from these studies. Is perlecan unique in its expression patterns, or are other BM components similar? How is tumor perlecan integrated into vascular BMs? Perlecan may self-associate through domain V (Yurchenco et al. 1987) but, equally, interactions of perlecan core protein and GAG chains with other macromolecules are possible. Do the tumor and host perlecans have different properties, such as the ability to bind growth factors? Are both required for tumor angiogenesis? Understanding of these issues may provide insights into the angiogenic process.
Downregulation of perlecan in tumor cells inhibits tumor growth and angiogenesis (Sharma et al. 1998; Ghiselli et al. 2001). Perlecan-deficient human colon carcinoma cells grow more slowly both in vitro and in vivo compared to the parental cell line. Unlike the parental cell line-generated tumor xenografts, which reveal extensive invasion of the deep fascia and subcutaneous skeletal muscles with abundant angiogenesis, the perlecan-deficient cells generated tumors with sharp boundaries, no infiltration of the deeper soft tissues, and no obvious angiogenesis (Ghiselli et al. 2001). Antisense targeting of perlecan in human colon carcinoma and mouse melanoma cells also caused substantial inhibition of tumor growth and angiogenensis (Sharma et al. 1998). To what extent is the inhibitory effect due to slowed proliferation rather than a lack of angiogenesis? Perhaps these are not completely separable, but they raise again the issue of growth factors that regulate both proliferation and angiogenesis.
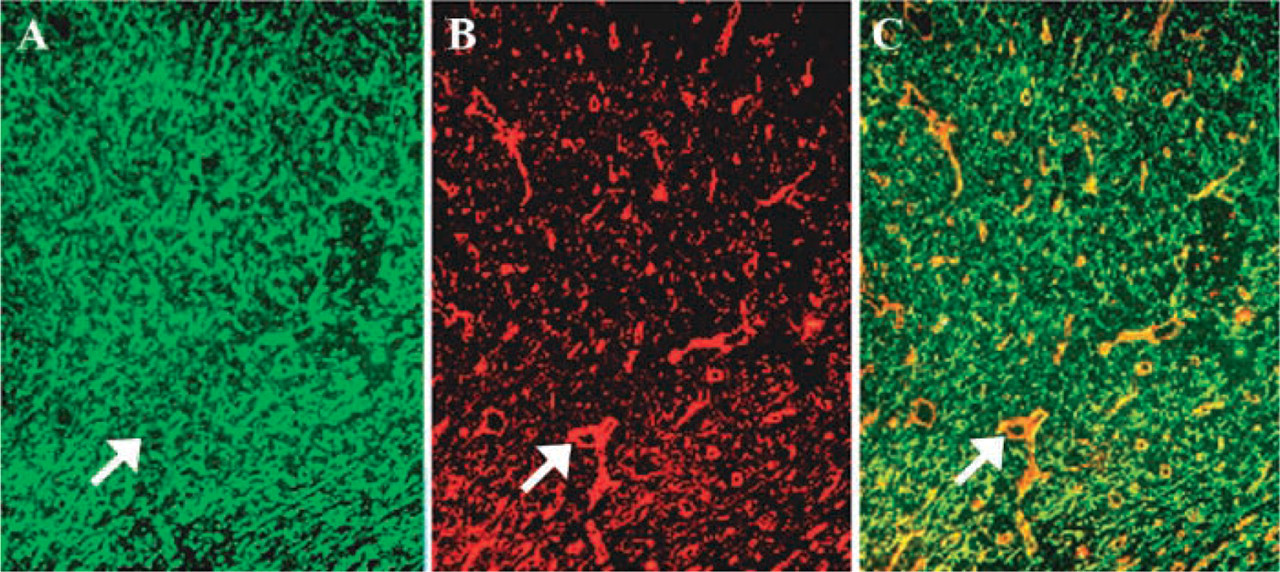
Localization of perlecan in an epidermal tumor. Frozen sections of a skin tumor generated by RT101 tumor cells were stained with rat anti-mouse perlecan antibody G9L1 (
Perlecan Interactions with Pro-angiogenic and Anti-angiogenic Regulators
Many pro-angiogenic regulators have been identified, including fibroblast growth factor (FGF)-1, −2, −3, −4, −5, and −7 (reviewed in Powers et al. 2000), vascular endothelial growth factor (VEGF), transforming growth factor (TGF)-α and -β, hepatocyte growth factor (HGF), tumor necrosis factor (TNF)-α, angiogenin, interleukin (IL)-8, angiopoietins (reviewed in Ferrara 1999), platelet-derived growth factor (PDGF) (reviewed in George 2001), and integrins αvβ3, αvβ5, and α5β1 (reviewed in Eliceiri and Cheresh 2001). Almost all of these, except integrins, interact with heparin and HS. The negative angiogenic regulators include thrombospondin (TSP)-1 and −2 (reviewed in Lawler 2000), endostatin, angiostatin, the 16-kD N-terminal fragment of prolactin, interferons (reviewed in Ferrara 1999), platelet factor-4 (PF-4) (Hagedorn et al. 2001), and tissue inhibitors of metalloproteinases (TIMPs; reviewed in Zetter 1998). Again, many of these bind heparin and HS. Here we focus on VEGF and FGF-2, two of the most potent and well-characterized heparin-binding angiogenic factors.
Vascular Endothelial Growth Factor
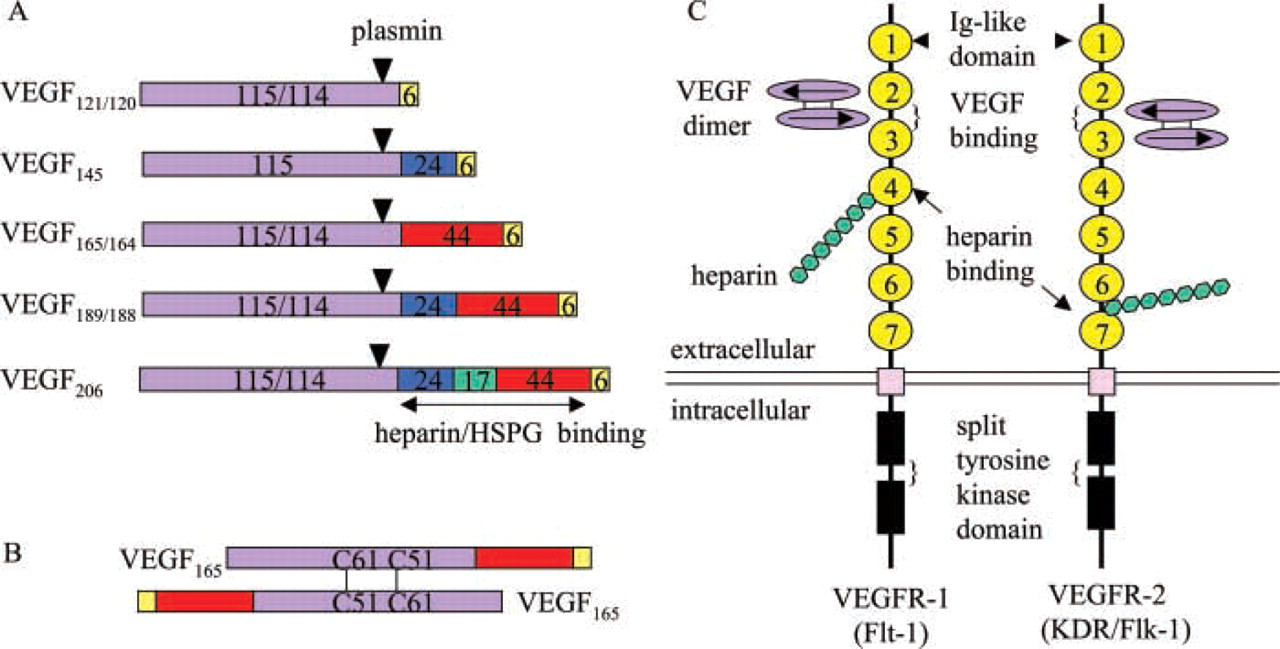
VEGF, a potent pro-angiogenic growth factor, stimulates the proliferation, migration, differentiation, and survival of endothelial cells. It was initially identified as vascular permeability factor (Senger et al. 1983). The VEGF family is composed of six members: VEGF-A or VEGF, placenta growth factor (PlGF), VEGF-B, -C, -D, and the orf parapox virus VEGF or VEGF-E (reviewed in Ferrara 1999). Alternative splicing of the VEGF gene results in five isoforms in human (VEGF121, VEGF145, VEGF165, VEGF189, VEGF206; Houck et al. 1991) and three isoforms in mice (VEGF120, VEGF164, VEGF188; Breier et al. 1992; Shima et al. 1996) (Figure 4). A potential N-linked glycosylation site exists at N74 of mouse or N75 of human VEGF (Breier et al. 1992). All VEGF isoforms are covalently linked homodimers. Each monomer contains cysteine-knot motif, and two antiparallel monomers are linked together by two intermolecular disulfide bonds formed between C51 and C61 (Muller et al. 1997) (Figure 4). VEGF165/164, a 34–46-kD glycoprotein, is the predominant isoform. Except for VEGF121/120, all other isoforms bind heparin with high affinity (Park et al. 1993). The expression of the VEGF gene is transcriptionally regulated by hypoxia, which occurs during tumor expansion, and ischemia (Minchenco et al. 1994). In mice, the loss of even a single VEGF allele results in embryonic lethality due to impaired angiogenesis and blood island formation (Carmeliet et al. 1996; Ferrara et al. 1996), indicating the irreplaceable function of VEGF in vasculogenesis.
Vascular Endothelial Growth Factor Receptors
The angiogenic activity of VEGF is mediated through two VEGF receptors (reviewed in Neufeld et al. 1999), VEGFR-1 or fms-like tyrosine kinase 1 (Flt-1) (de Vries et al. 1992) and VEGFR-2, known as kinase domain receptor (KDR) in human and fetal liver kinase 1 (Flk-1) in mice (Terman et al. 1992; Millauer et al. 1993). Both receptors are glycosylated and are characterized by the presence of seven Ig-like domains in the extracellular portion and a split TK domain intracellularly (Figure 4). Flt-1 and KDR/Flk-1 are mainly expressed on endothelial cells, and although the former exhibits higher binding affinity for VEGF (de Vries et al. 1992; Terman et al. 1992), it is KDR/Flt-1 that dominates VEGF induced mitogenic and angiogenic responses in endothelial cells (Walternberger et al. 1994; Keyt et al. 1996a,b; Gille et al. 2001). VEGFRs are essential for the development of the vascular system. Flk-1 null mice die by E9.5 due to an early defect in the development of endothelial and hematopoietic precursors. In these mice, blood island and blood vessels are absent (Shalaby et al. 1995). Flt-1-deficient mice die at E8.5 due to a failure of endothelial cells to assemble into an organized vascular system (Fong et al. 1995). Contrary to the absence of mature endothelial cells in Flk-1-null mice (Shalaby et al. 1995), the endothelial population in Flt-1 knockout mice is overcrowded due to increased mesenchymal-hemangioblast transition (Fong GH et al. 1999). Therefore, Flk-1 is essential for the development of endothelial cells, whereas Flt-1 is critical for hemangioblast commitment.
Schematic structures of VEGF isoforms and VEGFRs. (
Receptor Binding Sites on VEGF
The receptor binding sites on VEGF have been identified. VEGF165 can be cleaved by plasmin between residues R110 and A111. The resulting N-terminal fragment (residues 1–110) binds KDR and Flt-1 with almost the same affinity as VEGF165, whereas the C-terminal fragment (residues 111–165) binds heparin rather than KDR or Flt-1 (Keyt et al. 1996a). Therefore, The N-terminal part of VEGF165 is responsible for the TK receptor binding. Site-directed mutagenesis revealed that the negatively charged residues D63, E64, and E67 and the positively charged residues R82, L84, and H86 in VEGF165 are essential for binding Flt-1 and KDR, respectively (Keyt et al. 1996b). In VEGF164, amino acid residues 83–89 are required for Flk-1 binding. Substitution of this short peptide with the homologous region from human PlGF significantly reduces Flk-1 binding (Stacker et al. 1999). Detail regarding the receptor binding sites on VEGF comes from crystallographic data (Muller et al. 1997; Wiesmann et al. 1997). The crystal structure of human VEGF8-109 shows that two KDR-binding sites locate symmetrically at the opposite ends of the VEGF dimer (Muller et al. 1997). The crystal structure of VEGF8-109 in complex with Ig-like domain 2 of Flt-1 reveals that two copies of domain 2 bind to the opposite ends of VEGF dimer through a predominantly hydrophobic interaction, and the binding sites are very similar to that of KDR (Wiesmann et al. 1997).
Ligand-binding Sites on VEGF Receptors
The VEGF-binding sites on Flt-1 and KDR have been demonstrated by deletion analysis. Ig-like domains 2 and 3 of Flt-1 are necessary for high-affinity VEGF binding (Davis-Smyth et al. 1996; Wiesmann et al. 1997; Figure 4). Domain 2 by itself binds VEGF with a decreased affinity (about 60-fold) compared to the entire extracellular part of Flt-1 (Wiesmann et al. 1997), and deletion of domain 2 leads to complete abolition of VEGF binding (Davis-Smyth et al. 1996). Hence, domain 2 of Flt-1 is essential for VEGF binding. Domain 4 of Flt-1 is believed to be responsible for receptor dimerization (Barleon et al. 1997). Similar to Flt-1, Ig-like domains 2 and 3 of KDR are also sufficient for tight binding of VEGF (Fuh et al. 1998; Figure 4). Unlike to Flt-1, however, it is domain 3 rather than domain 2 that is critical for the binding of KDR to VEGF. Deletion of domain 3 completely abolishes VEGF binding. Domains 2 and 4 of KDR are important for ligand association, domains 5 and 6 are required for VEGF retention after binding, and domain 1 appears to have negative influence on VEGF binding (Shinkai et al. 1998). Other deletion experiments show that the first three Ig-like domains of Flt-1 and KDR are necessary for high-affinity VEGF binding (Barleon et al. 1997; Cunningham et al. 1997; Lu et al. 2000). The choice of domain boundaries may result in the contradictory data.
Heparan Sulfate Proteoglycans and VEGF Receptor Binding
The binding of VEGF to its receptors is modulated by heparin and cell surface HSPGs (Gitay-Goren et al. 1992,1993; Cohen et al. 1995; Gengrinovitch et al. 1999). Heparin strongly potentiates the binding of VEGF165 to its receptors on endothelial cells and melanoma cells. The binding is completely inhibited by heparinase digestion of the cells but can be restored by addition of exogenous heparin (Gitay-Goren et al. 1992, 1993), indicating that cell surface-associated HSPGs are required. To date, glypican-1 is the only cell surface HSPG that has been shown to enhance the binding of VEGF165 to its receptors (Gengrinovitch et al. 1999), although it is likely that other glypicans and syndecans share this role. The binding of glypican-1 to VEGF165 is mediated by the HS chains. Heparinase treatment reduces the binding of VEGF165 to its receptors on human vascular endothelial cells and addition of glypican-1 restores the binding. The ability of perlecan to potentiate the activity of VEGF has not been reported. Because perlecan is a pericellular HSPG, it is quite possible that it can modulate the activity of VEGF, and this will be important to ascertain.
In addition to VEGF, VEGFR1 and VEGFR2 are also heparin-binding molecules (Figure 4). The Ig-like domain 4 of Flt-1 is a major heparin binding site (Park and Lee 1999). A 10-amino-acid synthetic peptide between the Ig-like domains 6 and 7 of KDR binds heparin with high avidity (Dougher et al. 1997), and an Flk-1-alkaline phosphatase fusion protein also binds heparin (Chiang and Flanagan 1995). VEGF121, a non-heparin-binding VEGF isoform, requires cell surface HSPG for efficient binding. Predigestion of cells with heparinase inhibits the binding of VEGF121 to VEGFRs (Cohen et al. 1995), indicating that heparin and HSPG are important for the function of VEGFRs. VEGF, VEGFR, and cell surface HSPG may form a ternary complex that maximally potentiates the activity of VEGF. There is no report yet about the crystal structure of the ternary complex.
VEGF and Tumor Angiogenesis
The role of VEGF in tumor angiogenesis is under intense study. More than 1300 articles have been published in this field during the past 5 years. It is reported that many tumor cell lines can synthesize VEGF in vitro, such as mouse RT101 tumor cells (our unpublished data), human and rat prostate carcinoma cell lines (Chen et al. 2000), human glioma (Im et al. 1999), chondrosarcoma (Furumatsu et al. 2002), renal cell carcinoma (Shi and Siemann 2002), and pancreatic cancer cell lines (Luo et al. 2001). VEGF mRNA level is upregulated in a wide variety of human tumors, including breast, lung, kidney, gastrointestinal tract, bladder, and ovarian carcinomas (reviewed in Ferrara 1999), and the serum level of VEGF is increased in many cancer patients (Talks and Harris 2000). Overexpression of VEGF stimulates angiogenesis in human ovarian cancer xenografts (Duyndam et al. 2002) and enhances tumorigenicity of U251 MG glioma cells in vivo (Ke et al. 2002). Inhibition of tumor growth and angiogenesis in animal models has been demonstrated by neutralizing anti-VEGF antibody (Borgstrom et al. 1998), reduction of VEGF expression by antisense cDNA (Im et al. 1999; Luo et al. 2001) or antisense oligonucleotides (Shi and Siemann 2002), soluble Flt-1 (Goldman et al. 1998; Hoshida et al. 2002), dominant negative Flk-1 (Millauer et al. 1994), anti-KDR antibody (Sweeney et al. 2002), Flk-1/KDR kinase inhibitor SU5416 (Fong TA et al. 1999; Takamoto et al. 2001), anti-VEGF189 or anti-Flk-1 or Flt-1 ribozymes (Oshika et al. 2000; Pavco et al. 2000), and cytotoxic T-lymphocytes engineered to target Flk-1 (Niederman et al. 2002). A synthetic peptide that blocks VEGF-KDR interaction or derives from the second Ig-like domain of Flt-1 can abolish angiogenesis in rabbit corneal and chick chorioallantoic membrane angiogenesis models, respectively (Binetruy-Tournaire et al. 2000; Tan et al. 2001). However, the effect of such peptides on tumor angiogenesis in vivo has not been revealed thus far. Lu et al. (2001) reported that a bifunctional antibody (“diabody”) directed against Flt-1 and Flk-1 strongly inhibits the mitogenic response of human endothelial cells to VEGF, which appears to offer an opportunity to block tumor angiogenesis in vivo.
Drosophila VEGFs and VEGFRs
Recently, both VEGF and VEGFR homologues have been found in Drosophila (Cho et al. 2002). The three VEGF homologues are encoded by three genes, whereas the three VEGFR homologues are produced by alternative splicing of a single gene. VEGFR homologues are expressed in hemocytes, and a VEGFR mutation causes the disruption of blood cell migration during embryonic development. VEGF homologues are expressed along blood cell migration routes. Simultaneous inactivation of all three VEGF genes causes blood cell migration defects, and ectopic expression of VEGF redirects the migration. These results suggest that the ancestral function of VEGF is to control blood cell migration. With vertebrate evolution, VEGF has become associated with more complicated processes, such as vasculogenesis and angiogenesis.
Fibroblast Growth Factor-2
The FGF family contains at least 20 members, which are structurally related heparin-binding proteins. FGF-2, also known as basic FGF (bFGF), was the first pro-angiogenic growth factor to be identified (Shing et al. 1984). Similar to VEGF, FGF-2 also stimulates the proliferation, migration, differentiation, and survival of endothelial cells. Alternative translation of FGF-2 mRNA results in four isoforms with molecular masses of 18, 22, 22.5, and 24.2 kD, respectively (Figure 5). The 18-kD form is translated from the start codon AUG. The others are translated from the upstream and in-frame CUG codons (Florkiewicz and Sommer 1989; Prats et al. 1989). Unlike VEGF and other growth factors, which are homodimeric and are secreted by cells, FGF-2 is monomeric and lacks a signal sequence for secretion (reviewed in Wiesmann et al. 2000). The 18-kD form is localized primarily in the cytoplasm, whereas the larger forms are transported to nucleus (Bugler et al. 1991). The mechanism of FGF-2 release remains unclear. FGF-2-null mice are viable. The newborn mice display defects in organization and differentiation of the cerebral cortex, which is compressed compared to wild-type mice. Adult mice show reduced blood pressure and delayed wound healing (Dono et al. 1998; Ortega et al. 1998). The mild phenotype of FGF-2-null mice suggests redundancy in the FGF family.
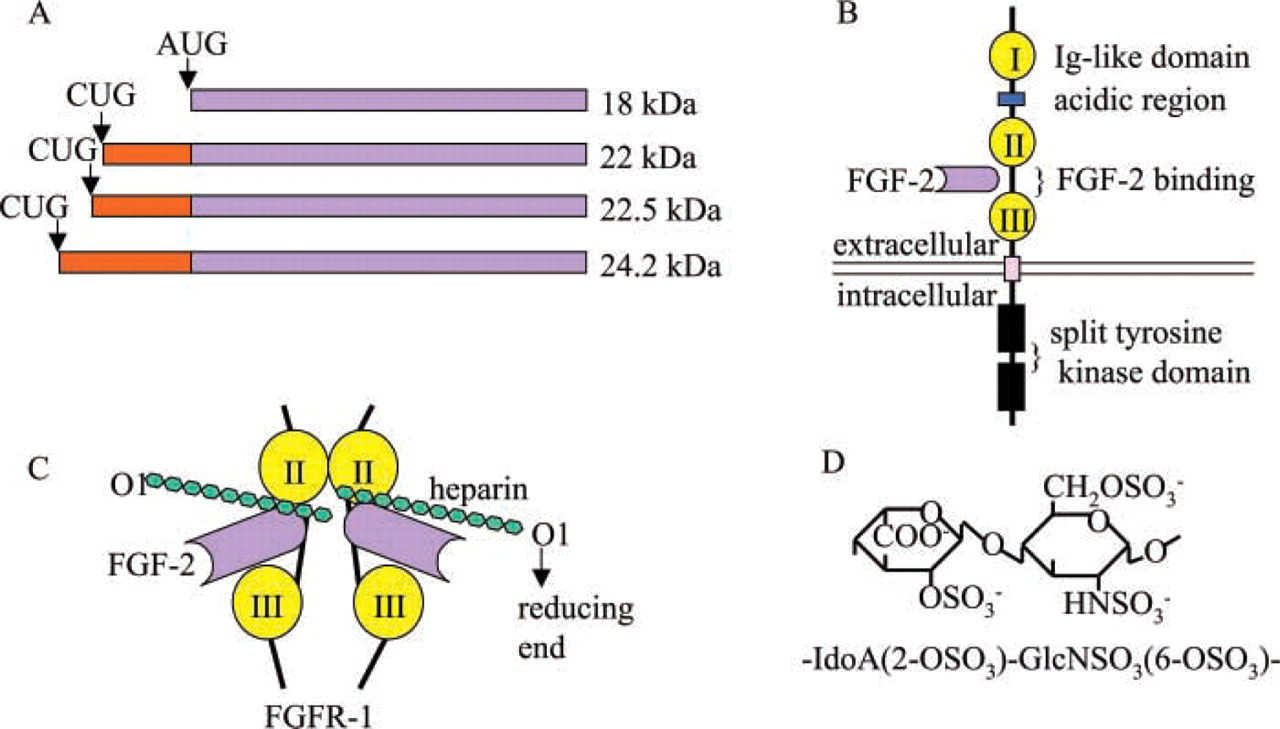
Schematic structures of FGF-2 and FGFRs. (
Fibroblast Growth Factor Receptors
The biological effects of FGF-2 are mediated by high-affinity FGF receptors (FGFR-1, −2, −3, and −4) and low-affinity receptors (Klagsbrun and Baird 1991). FGFRs are characterized by the presence of three Ig-like domains in the extracellular part and a split TK domain intracellularly. An acidic region, which consists of an acidic stretch of approximately 30 residues, is located between Ig-like domains I and II (Figure 5). The Ig-like domain III is encoded by either exons IIIa and IIIb or IIIa and IIIc. The choice of IIIb or IIIc determines the ligand binding specificity of the receptor. For example, FGF-7 only binds FGFR-2(IIIb) but not FGFR-2(IIIc). The other domains of FGFRs also undergo alternative splicing, which increases the diversity of FGFRs. The expression of FGFR isoforms is cell type-specific, e.g., FGFR-2 (IIIb) and 2(IIIc) are restricted to cells of epithelial and mesenchymal lineages, respectively. Compared to FGFR-1, −2, and −3, FGFR-4 is unusual because it is not subject to alternative splicing (reviewed in Powers et al. 2000).
RNA in situ hybridization studies revealed that each FGFR has a unique expression pattern during vertebrate embryogenesis. FGFR-1 and FGFR-2 are co-expressed in some tissues, such as the mesoderm and neuroectoderm. However, FGFR-1 is expressed almost exclusively in mesenchyme (Orr-Urtreger et al. 1991; Peters et al. 1992; Yamaguchi et al. 1992), whereas FGFR-2 is expressed predominantly in epithelium (Orr-Urtreger et al. 1991,1993; Peters et al. 1992). FGFR-3 is expressed in the developing CNS and the cartilage rudiments of developing bones (Peters et al. 1993), while FGFR-4 is expressed in the definitive endoderm, the somatic myotome and the myotomally derived skeletal muscle (Stark et al. 1991).
The importance of each receptor during murine development is revealed by gene targeting experiments. FGFR-1-deficient embryos display severe growth retardation and aberrant mesodermal patterning; all embryos die before or during gastrulation (Deng et al. 1994; Yamaguchi et al. 1994). The function of FGFR-1 in later stages of embryogenesis is revealed by chimeras generated by injecting FGFR-1-deficient ES cells into wild-type blastocysts. The results show that embryos with a low contribution of deficient cells can complete gastrulation but display abnormalities at later stages, including spina bifida, partial duplication of the neural tube, tail distortion, limb bud malformation, and truncation of embryonic structures (Ciruna et al. 1997; Deng et al. 1997). Mice carrying a targeted deletion of the transmembrane and part of the tyrosine kinase domain of FGFR-2 die at E4.5–5.5 (Arman et al. 1998), and those carrying a targeted deletion of the entire Ig-like domain III of FGFR-2 die at E10-11, with a failure to form functional placenta and limb buds (Xu et al. 1998). Disruption of the gene encoding mouse FGFR-3 leads to skeletal and inner ear defects. However, the homozygous mutants can survive (Colvin et al. 1996; Deng et al. 1996). Mice homozygous for a targeted mutation of FGFR-4 exhibit no overt abnormalities except that the cholesterol metabolism and bile acid synthesis are increased (Weinstein et al. 1998; Yu et al. 2000).
Heparan Sulfate Proteoglycans and FGF-2
Heparin or cell surface HSPG functions as a low-affinity FGF receptor, which is necessary for the binding of FGF-2 to high-affinity FGFRs. FGFR-expressing HS-deficient CHO mutant cell lines do not bind FGF-2 unless exogenous heparin or HS is added (Yayon et al. 1991). It is proposed that the activation of monomeric FGFs occurs as a result of their association as dimers or oligomers on heparin or HS-chain (reviewed in Gallagher 2001). This proposition is supported by the crystal structure of a ternary complex formed by FGF-2, FGFR-1, and heparin (Schlessinger et al. 2000) (Figure 5). In this complex, FGF-2 and Ig-like domains II and III of FGFR-1 form a 2:2 tetrameric assembly with each of the FGF-2:FGFR-1 dimers in association with separate decasaccharides. Within each dimer, FGF-2 interacts extensively with Ig-like domains II and III as well as the linker between the two domains. The two heparin decasaccharides are arranged in an antiparallel fashion and make extensive contacts with a canyon of positive charge that extends across the ligand receptor pairs (Plotnikov et al. 1999, Schlessinger et al. 2000). The crystal structure of FGF-1: FGFR-2:heparin ternary complex reveals a different stoichiometry of heparin binding in the complex (Pellegrini et al. 2000). Here, FGF-1 and the Ig-like domains II and III of FGFR-2 also form a 2:2 tetramer, and FGF-1 is bound at the junction between the two domains of one FGFR-2. However, only a single asymmetric heparin decasaccharide is present in the complex, the decasaccharide interacts with both FGF-1 monomers but only one FGFR-2 molecule. The ternary complex is assembled around a central heparin molecule that links two FGF-1 monomers into a dimer, which bridges between two FGFR-2 molecules (Pellegrini et al. 2000; Stauber et al. 2000).
The ability of perlecan to modulate FGF-2 activity has been well studied. Perlecan potentiates the high affinity binding of FGF-2 to HS-deficient CHO cells and to soluble FGFRs. It also strongly augments the mitogenic activity of FGF-2 and induces FGF-2 mediated angiogenesis in vivo (Aviezer et al. 1994). Reduced expression of perlecan by antisense cDNA decreases the binding and mitogenic response of mouse NIH 3T3 cells and human metastatic melanoma cells to FGF-2, and the phenotype can be rescued by exogenous heparin or perlecan (Aviezer et al. 1997). The ability of perlecan to bind FGF-2 depends on the primary sequence of HS chains that varies in different cell types (Whitelock et al. 1999; Knox et al. 2002). 2-O-sulfated iduronic acid and N-sulfated glucosamine have been shown to be required for the high affinity binding between FGF-2 and HSPG (Turnbull et al. 1992) (Figure 5).
FGF-2 and Tumor Angiogenesis
FGF-2 bound to perlecan HS-chains can be released very efficiently by stromelysin, collagenase, and plasmin, which degrade the perlecan core protein (Whitelock et al. 1996), suggesting that perlecan can act as a store of FGFs. FGF-2 released by proteolytic enzymes may play an active role in tumor growth and angiogenesis. Many tumor cell lines, including mouse RT101 tumor cell line (our unpublished data), human chondrosarcoma (Furumatsu et al. 2002), melanoma (Wang and Becker 1997), transitional cell carcinoma (TCC) of the bladder (Inoue et al. 2000), and HT-29 human colon cancer cell lines (Netzer et al. 2001), can synthesize FGF-2. Overexpression of FGF-2 in low metastatic renal carcinoma cells results in increased angiogenesis and metastatic potential in vivo (Miyake et al. 1996). FGF-2 levels in the serum of patients with breast cancer (Sluitz et al. 1995), colorectal cancer (Landriscina et al. 1998), renal cell carcinoma (Dosquet et al. 1997), and other cancers are inversely correlated with survival. Downregulation of FGF-2 by antisense cDNA (Wang and Becker 1997; Inoue et al. 2000) or antisense oligonucleotides (Netzer et al. 2001), neutralizing anti-FGF-2 antibody (Aonuma et al. 1999), peptide corresponding to the heparin-binding domain of FGF-2 (Plum et al. 2001), and reduced expression of FGFR-1 by antisense cDNA (Wang and Becker 1997) or antisense oligonucleotides (Yamada et al. 1999) have been shown to block tumor growth and angiogenesis in vivo.
Synergy Between VEGF and FGF-2
As the two most common pro-angiogenic growth factors, VEGF and FGF-2 can act synergistically both in vivo and in vitro. FGF-2 induces VEGF gene expression in endothelial cells, and neutralizing anti-VEGF antibody inhibits FGF-2 induced endothelial cell proliferation in vitro and angiogenesis in a mouse corneal pocket angiogenesis model (Seghezzi et al. 1998). In a rat sponge implant angiogenesis model in which subcutaneous sponge disks are implanted daily, injection of FGF-2 into the sponge implants enhances VEGF expression and angiogenesis in the granuloma tissue developed around the implants, while VEGF antisense oligonucleotides significantly inhibit FGF-2 induced angiogenesis (Majima et al. 2000). FGF-2 also induces the expression of VEGFR-2 in endothelial cells (Pepper and Mandriota 1998; Hata et al. 1999). Overexpression of both VEGF and FGF-2 in murine HCC cells synergistically increased tumor growth and angiogenesis in vivo, whereas neutralizing anti-KDR/Flk-1 antibody significantly suppressed FGF-2-induced tumor growth (Yoshiji et al. 2002). These results suggest a possible mechanism underlying the synergy between FGF-2 and VEGF. FGF-2 induces both VEGF and VEGFR-2 expression, and the increased VEGFR-2 expression can further potentiate the activity of VEGF, leading to additional VEGFR-2 expression. The positive feedback loop results in significantly increased endothelial cell proliferation and angiogenesis.
Other Perlecan-binding Angiogenic Regulators
In addition to FGF-2, perlecan can bind a number of other pro-angiogenic and anti-angiogenic regulators through its HS-chains or core protein. FGF-1, −4, −7 (Tapanadechopone et al. 2001), HGF, TGF-β, PF-4 (reviewed in Iozzo and San Antonio 2001), and TSP-1 (Vischer et al. 1997) can bind to the HS-chains. The binding sites for FGF-7 and PDGF are located on domain III-1 and III-2 of perlecan core protein, respectively (Gohring et al. 1998; Mongiat et al. 2000). Perlecan may modulate the activities of these regulators by either sequestering them in the BMs and ECM or presenting them to their cell surface high-affinity receptors.
Conclusions
It has been shown that perlecan can support integrin-mediated adhesion (Hayashi et al. 1992; Battaglia et al. 1993; Brown et al. 1997; Whitelock et al. 1999), a perhaps vital part of the migrating process that underlies both tumor invasion and endothelial cell migration in angiogenesis. Given the complex interactions of perlecan as a matrix molecule and its potential to serve as a repository for angiogenic growth factors, it is not surprising that much remains unresolved. Perlecan is unique in its structure, is a single family member, yet is indispensable for invertebrate and vertebrate life. To counterbalance this, perlecan expression may augment the growth and survival of a range of tumor types, in part by promoting some of the same vascular effects that are required in cardiovascular development. The connection between perlecan's HS-chains and angiogenic growth factors seems certain and is likely to remain a major research focus. It would be revealing to uncover the molecular basis by which some tumors are promoted by the presence of perlecan, while others are inhibited. With such detailed understanding it may become possible to intervene in the unwanted side of perlecan's properties.
Footnotes
Acknowledgements
Supported in part by National Institutes of Health Grant AR36457 to JRC.
We thank Dr. Paraith Tapanadechopone for the pioneering work of immunofluorescence staining of the RT101 tumor.