
Abstract
As a major heparan sulfate proteoglycan (PG) in basement membranes, perlecan has been linked to tumor invasion, metastasis, and angiogenesis. Here we produced epidermal tumors in immunocompromised rats by injection of mouse RT101 tumor cells. Tumor sections stained with species-specific perlecan antibodies, together with immuno-electron microscopy, showed that perlecan distributed around blood vessels was of both host and tumor cell origin. Tumor-derived perlecan was also distributed throughout the tumor matrix. Blood vessels stained with rat-specific PECAM-1 antibody showed their host origin. RT101 cells also expressed two other basement membrane heparan sulfate PGs, agrin and type XVIII collagen. Antisense targeting of perlecan inhibited tumor cell growth in vitro, while exogenous recombinant perlecan, but not heparin, restored the growth of antisense perlecan-expressing cells, suggesting that perlecan core protein, rather than heparan sulfate chains from perlecan, agrin, or type XVIII collagen, regulates tumor cell growth. However, perlecan core protein requirement was not related to fibroblast growth factor-7 binding because RT101 cells were unresponsive to and lacked receptors for this growth factor. In vivo, antisense perlecan-transfected cells generated no tumors, whereas untransfected and vector-transfected cells formed tumors with obvious neovascularization, suggesting that tumor perlecan rather than host perlecan controls tumor growth and angiogenesis.
Keywords
P
Perlecan has been linked to tumor invasion, metastasis, and angiogenesis. In human metastatic melanomas, perlecan mRNA level is markedly increased compared with normal tissue. The overexpressed perlecan is deposited in the pericellular matrix and appears to be related to the invasiveness of the melanoma cells (Cohen et al. 1994). In human breast (Guelstein et al. 1993), liver (Roskams et al. 1998), ovarian (Gonzalez et al. 2003), colon, and prostate carcinomas (reviewed in Iozzo et al. 1994), high levels of perlecan are detected around newly formed blood vessels. Antisense reduction of perlecan reduces proliferation and invasiveness of murine melanoma cells and causes significant inhibition of tumor growth and angiogenensis in tumor xenografts induced by human colon carcinoma and mouse melanoma cells (Adatia et al. 1997; Sharma et al. 1998). Perlecan-deficient human colon carcinoma cells also show remarkably decreased growth both in vitro and in vivo, and no obvious angiogenesis is observed in tumors induced by perlecan-deficient cells (Ghiselli et al. 2001). Interestingly, perlecan by itself is able to stimulate angiogenesis in a rabbit ear model (Aviezer et al. 1994). Therefore, perlecan may provide a scaffold for tumor angiogenesis. Conversely, inhibitory effects of perlecan on tumor growth and angiogenesis have been revealed in other studies. Recently, endorepellin, which is derived from the C-terminal domain V of perlecan, has been shown to inhibit endothelial cell migration, endothelial tube-like formation within type I collagen matrix, and angiogenesis in chicken chorioallantoic membrane and Matrigel plug assays (Mongiat et al. 2003), indicating that perlecan may have an anti-angiogenic effect in vivo if domain V is released from perlecan through protease cleavage. In human fibrosarcoma (Mathiak et al. 1997) cells, antisense suppression of perlecan increases cell migration and invasion of ECM. In human Kaposi's sarcoma cells (Marchisone et al. 2000), antisense targeting of perlecan results in stimulation of tumor cell growth in vivo. In these cases, lack of perlecan may favor the diffusion of heparinbinding growth factors, leading to enhanced tumor growth.
In the process of tumor growth, both tumor and host cells may synthesize perlecan. Data from Iozzo et al. (1994) indicated that perlecan deposited in the BMs of newly formed blood vessels in tumors was of tumor origin. We investigated mouse tumors produced in immunocompromised rats by intradermal injection of mouse RT101 tumor cells to determine the specific origin of perlecan, the distribution of type IV collagen, laminin, and entactin/nidogen-1. Furthermore, we characterized the effect of perlecan reduction in tumor cells on tumor growth and neovascularization.
Materials and Methods
Cells
The RT101 cell line was kindly provided by Dr. N.H. Colburn (National Cancer Institute, National Institutes of Health, Bethesda, MD) through Drs. P Chang and C Prince (University of Alabama at Birmingham, AL). The RT101 cell line was generated by irreversible transformation of JB6 Cl 41.5a, a mouse epidermal cell line, with phorbol ester treatment. RT101 cells were grown in Eagle's minimal essential medium (Invitrogen; Paisley, UK) supplemented with 5% heat-inactivated fetal calf serum at 37C with 10% CO2. Cells were subcultured every 2 or 3 days. Drosophila S2 cells (Invitrogen; Carlsbad, CA) were maintained in complete DES Expression Medium (Invitrogen) containing 10% heat-inactivated fetal calf serum (FCS) and glutamine at 22–24C without CO2. Medium was replaced every 3 or 4 days.
Primary Antibodies
All primary antibodies used in this study are summarized in Table 1.
Primary antibodies a
aIF, immunofluorescence; IB, immunoblotting; ImmunoEM, immunoelectron microscopy.
Analysis of PGs Prepared from Conditioned Medium
Serum-free medium conditioned by confluent RT101 cells for 24 hr was collected and centrifuged at 1000 rpm for 5 min to remove cell debris. The supernatant was applied on a 3 × 5-cm DEAE-Sephacel (Amersham Bioscience; Piscataway, NJ) column equilibrated with 50 mM Tris-HCl buffer (pH 8.0) containing 4 M urea, 0.2 M NaCl, 10 mM NEM, 20 mM EDTA, 0.2 mM PMSF, and 0.1% Tween-20. The column was washed with 50 mM sodium acetate buffer (pH 4.0) containing the same components as the 50 mM Tris-HCl buffer, followed by another wash with the same sodium acetate buffer but in the absence of urea. PGs were subsequently eluted with 4 M guanidine-HCl, pH 4.0. containing 50 mM sodium acetate and 0.1% Tween-20. The protein-containing fractions were collected and precipitated by adding 4 or 5 volumes of ice-cold ethanol. The mixture was incubated at −20C for at least 3 hr. The precipitated pellet was air-dried and resuspended in heparinase buffer (0.1 M sodium acetate, 0.1 mM calcium acetate, pH 7.0).
Aliquots of 20 μl of PG preparations were treated with 1 mU of heparinase III (EC 4.2.2.8) and/or 5 mU of chondroitinase ABC (EC 4.2.2.4; Seikagaku America, Falmouth, MA) in the presence of 10 μg/ml ovomucoid (Sigma; St Louis, MO). After incubation at 37C for 6 hr or overnight, the samples were resolved by 3–15% SDS-PAGE gel, followed by membrane transfer. Membranes were blocked with PBS (for monoclonal antibodies) or TBS (for polyclonal antibodies) containing 5% skimmed milk and 0.1% Tween-20 at room temperature for 1 hr, followed by primary antibody incubation. Biotin-conjugated HS stub antibody F69-3G10, perlecan antibody H5L5, and polyclonal agrin and type XVIII collagen antibodies were used as summarized in Table 1. Monoclonal antibodies were diluted with PBS containing 1% skimmed milk and 0.1% Tween-20 (MPBS/T), while polyclonal antibodies were diluted with TBS containing 1% skimmed milk and 0.1% Tween-20 (MTBS/T). The membranes were incubated with primary antibodies at RT for 1 to 2 hr or overnight at 4C. After washing with MTBS/T or MPBS/T, membranes were incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (swine anti-rabbit IgG, rabbit anti-mouse IgG, rabbit anti-rat IgG, all diluted 1:5000; DAKO, Ely, Cambridgeshire, UK) at RT for 1 hr, washed, and developed with enhanced chemiluminescence (ECL) reagents (Amersham Bio-sciences).
cDNA Constructs
A 5.0-kb cDNA fragment encoding mouse perlecan domain I/II/III with a 5′-end perlecan signal sequence was released from the original vector pBS (a gift from Dr. John R. Hassell; Shriners Hospital for Children, Tampa, FL) with Not I and Xba I and inserted into vector pMT/V5-His A (Invitrogen) with the same enzymes.
A cDNA fragment encoding mouse perlecan domain I, II, and 11 amino acids of domain III (nucleotides 579-2118, GenBank accession no. M77174) was amplified from perlecan construct I/II/III-pBS by PCR. The upstream and downstream primers were 5′-TATATAATGGGGCAGCGGGCA-3′ and 5′-CCAAGCTTCCAGGTAGAAGTG-3′, respectively. A restriction site for Hind III (underlined) was introduced into the downstream primer. For PCR amplification, 30 cycles of 95C for 30 sec, 58C for 1 min, and 72C for 2 min, with a final extension at 72C for 7 min, were performed. The PCR product was cloned into prelinearized pCR2.1 vector using TOPO TA cloning system (Invitrogen; Paisley, UK). This cDNA fragment (1.54 kb) was inserted in an antisense orientation into vector pcDNA3.1/HisA (Invitrogen) between Hind III and EcoR I sites. The antisense orientation of perlecan on vector pcDNA3.1/HisA was confirmed by sequencing.
Mouse perlecan construct I/II/III-pRC/CMV was a gift from Dr. John R. Hassell. The cDNA fragment encoding part of perlecan domain I (nucleotides 579-1087) was amplified from construct I/II/III-pRC/CMV by PCR using upstream primer (T7) (5′-TAATACGACTCACTATAGGG-3′) and downstream primer (5′-GCCTCTAGAGACCACTGTATG-3′). An Xba I site (underlined) was introduced into the downstream primer. Thirty PCR cycles of 95C for 30 sec, 52C for 30 sec, and 72C for 1 min, with a final extension at 72C for 7 min, were performed. The PCR product was purified using PCR purification kit (QIAGEN; Crawley, UK), digested with Not I and Xba I, and inserted into pRC/CMV vector with the same enzymes.
Stable Transfection of RT101 Cells
RT101 cells were plated into T-25 flasks 16 hr before transfection. Serum-free medium (300 μl) containing 5 μg of empty vector or antisense perlecan I/II-pcDNA3.1/HisA DNA was mixed with 300 μl of serum-free medium containing 20 μl of Lipofectamine (Invitrogen), incubated at RT for 45 min, added to RT101 cells (50% confluence), and incubated with RT101 cells for 6 hr. The cells were washed and maintained in serumcontaining medium at 37C, 10% CO2 for 2 days, followed by G-418 (750 μg/ml; Invitrogen) selection. The resistant cells were expanded by ring cloning, and perlecan levels in antisense clones were analyzed by dot-blot and cell staining.
To perform dot-blots, antisense clones, vector-transfected, or untransfected cells were grown in 6-well plates (Fisher; Loughborough, UK) until reaching confluence. Cells were washed twice with serum-free medium and maintained in serum-free medium at 37C, 10% CO2 for 24 hr. The media were collected and centrifuged at 1000 rpm for 5 min to remove cell debris. The supernatants were loaded onto PVP membrane (pretreated with methanol) in a dot-blot apparatus (BioRad; Hemel, UK). Supernatant loading amounts were normalized by cell number. Then 1 × and 4 × loading were performed for each sample. The membrane was washed with TBS/T, followed by immunoblotting with perlecan antibody R63.
To perform immunofluorescence staining, untransfected, vector-transfected, or antisense-perlecan expressing cells grown on coverslips for 4 days were rinsed with warm PBS and fixed with warm freshly made paraformaldehyde containing 0.1% Triton X-100 at RT for 20 min. Cells were washed with PBS, incubated with 0.1 M NH4Cl/PBS at RT for 15 min, washed with PBS, and incubated with perlecan antibody A7L6 at 37C for 1 hr. After washing with PBS, the coverslips were incubated with fluorescein isothiocyanate (FITC)-conjugated cross-absorbed goat anti-rat IgG (diluted 1:100; Jackson ImmunoResearch, Soham, UK) at 37C for 1 hr. The cells were then washed, mounted, and observed by epifluorescence microscopy.
To analyze the growth rate of antisense clones in vitro, antisense-perlecan transfected, vector-transfected, or untransfected cells were seeded into 24-well plates (5 × 104 cell/well) and grown in serum-containing medium at 37C, 10% CO2 for 1, 2, or 3 days, after which cell numbers were counted. To test the effects of heparin (Sigma; Poole, UK) or recombinant perlecan on tumor cell growth, 5 × 104 cells (for recombinant perlecan) or 1 × 104 cells (for heparin) were seeded into 24-well plates and grown at 37C, 10% CO2 for 3 days in 0.1% FCS containing medium in the presence of 1 μg/ml heparin, 10 μg/ml heparin, or 10 μg/ml recombinant perlecan. All samples were tested in triplicate and the experiments were repeated twice.
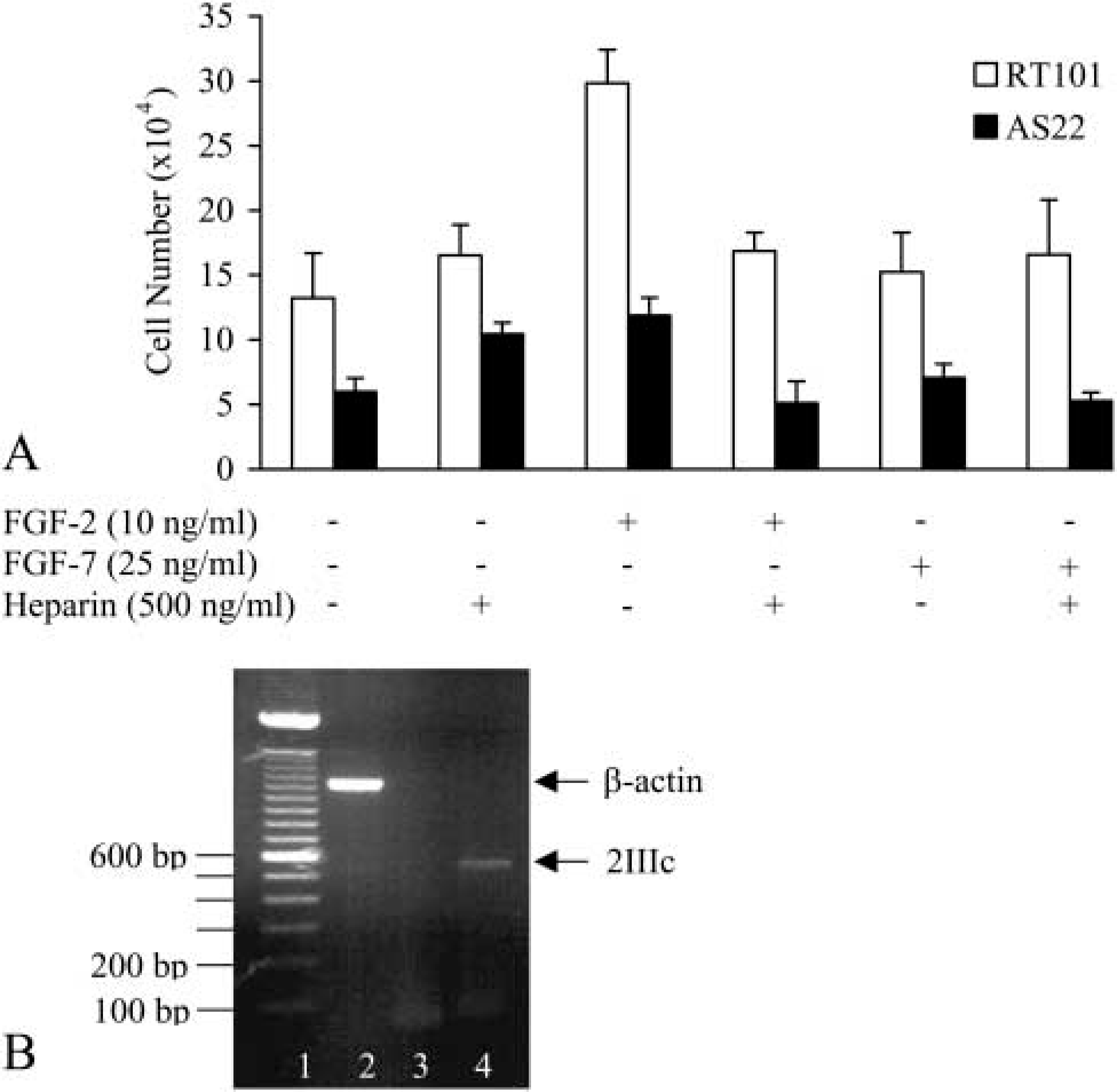
RT101 cells produce fibroblast growth factor (FGF)-2 and −7. To examine the effects of FGF-2 and −7 on RT101 cells, 5 × 104 untransfected or antisense-perlecan transfected cells were seeded into 24-well plates and grown in 0.1% FCS-containing medium for 24 hr (37C, 10% CO2) in the absence or presence of FGF-2 (10 ng/ml; R and D Systems, Abingdon, UK), FGF-7 (25 ng/ml; R and D Systems), or heparin (500 ng/ml). Each treatment was performed in quadruplicate and repeated twice.
RT-PCR
Total RNA was prepared from RT101 cells in culture. Cells were extracted from a T75 flask and spun down at 900 rpm for 5 min at RT. The supernatant was removed and the cells were lysed with 1 ml RNAzol B reagent (Leedo Medical Laboratories; Houston, TX). Total RNA was extracted by chloroform and precipitated by isopropanol. RNA was pelleted by centrifugation at 12,000 × g for 15 min at 4C, washed twice with 200 μl of ice-cold ethanol, air-dried, and resuspended in nuclease-free water and stored at −80C.
The expression of FGF receptor (FGFR)-2 on RT101 cells was analyzed by RT-PCR using a Reverse Transcription System (Promega; Southampton, UK). For FGFR-2IIIb, the primer sequences were 5′-AACACTGTGAAGTTCCGCTG-3′ (nucleotides 847-866, GenBank accession no. M63503) and 5′-AGGCAGACTGGTTGGCCTG-3′ (nucleotides 1372-1390). For FGFR-2IIIc, the upstream primer sequence was the same as that of isoform IIIb and the downstream primer was 5′-TGGCAGAACTGTCAACCATG-3′ (nucleotides 1670-1689, GenBank accession no. NM_010207). One μg of total RNA was used in each reaction. Reverse transcription was carried out at 42C for 40 min. The product was diluted to a final volume of 100 μl with nuclease-free water. For amplification of FGFR-2, 20 μl of the first-strand cDNA was added to the reaction mixture (50 μl). The reaction was performed at 94C for 3 min, followed by 35 cycles of 94C for 45 sec, 62C for 45 sec, and 72C for 1.5 min with a final extension at 72C for 10 min. Amplification of actin cRNA was carried out as a positive control. The products were analyzed on a 1% agarose gel.
Tumor Production and Processing
Approximately 3 × 106 RT101 cells were injected intradermally into 6-8-week-old male immunocompromised NIHRNU-M rats (Taconic Farms; Germantown, NY). Two weeks later, tumors were harvested and either processed for electron microscopy (EM) or snap-frozen in liquid nitrogen and stored at −80C for frozen-section and Western blotting. Animal care and use procedures were fully in accordance with all national and local requirements. Animals were under the care of full-time accredited veterinarians.
To prepare samples for EM, small tumor pieces were washed, fixed, dehydrated, and embedded as described by Glauert (1975). Semithin sections (1 μm) were stained with 1% toluidine blue in 1% sodium borate. For conventional EM, ultrathin sections (60-90 nm) were stained with 3% aqueous uranyl acetate and Reynolds' lead citrate for 10 min each.
To determine the origin(s) of perlecan in tumors, frozen-sections (6 μm) were washed three times with PBS at RT, treated with PBS containing 1% NH4Cl/PBS at RT for 15 min, blocked with 1% BSA/PBS at 37C for 1 hr, and incubated with primary antibodies at 37C for 1 hr. Perlecan antibodies 11B4 and G9L1, PECAM-1 antibodies MEC13.3 and TLD-3A12, entactin/nidogen-1 antibody 10C7, laminin antibody D18, laminin-5 antibody J18, and type IV collagen antibody were used, as summarized in Table 1. At the end of primary antibody incubation, tumor sections were washed with PBS, incubated with appropriate FITC-conjugated swine anti-rabbit IgG (diluted 1:40; DAKO), Texas Red (TR)-conjugated goat anti-mouse IgG (cross-absorbed, diluted 1:1000; Molecular Probes, Leiden, The Netherlands), or Alexa Fluor 568-conjugated goat anti-mouse IgG (cross-absorbed, diluted 1:1000; Molecular Probes) at 37C for 1 hr, washed with PBS, mounted, sealed, and observed under epifluorescence optics. Tumor sections incubated with secondary antibodies only were used as negative controls. Species-specificity of the monoclonal antibodies was confirmed by fluorescence staining of mouse and rat kidney sections. All of the controls showed the expected staining patterns, with no inappropriate crossreactivity.
To analyze the distribution of tumor and host perlecan in more detail, immunoelectron microscopy (immunoEM) was performed. Tumor ultrathin sections were mounted on Formvar-coated nickel grids. Nonspecific binding sites were blocked by floating the grids on drops of PBS containing 1% BSA and 0.01% Tween-20 for 15 min. Sections were incubated overnight at 4C with perlecan antibodies G9L1 and 11B4 diluted in PBS containing 0.1% BSA and 0.01% Tween-20 (secondary antibodies were diluted in the same solution), followed by incubation with 10-nm gold-conjugated goat anti-rat antibodies (diluted 1:20; E-Y Laboratories, San Mateo, CA) and 15-nm gold-conjugated rabbit anti-mouse antibodies (diluted 1:20; E-Y Laboratories) for 1 hr. As controls, primary antibodies were omitted, or either one of the two primary antibodies was used, and detected using a mixture of both secondary antibodies. Sections were then washed and counterstained with 3% aqueous uranyl acetate and Reynolds' lead citrate, 2 min each. The sections were examined in a JEOL JEM-1200 EX microscope.
To investigate the effect of perlecan reduction on tumor growth and neovascularization in vivo, 1 × 106 untransfected, vector-transfected, or antisense perlecan-transfected cells were injected intradermally into 6-8-week-old NIHRNU-M rats (Charles River Wiga Deutschland; Kisslegg, Germany). To reduce individual differences, contralateral injections were performed. The rats were divided into two groups with five rats in each group. Five weeks later, tumors were harvested, weighed, and processed for frozen sections or paraffin sections.
To examine perlecan mRNA in tumors, in situ hybridization (ISH) was performed as described by Schaefer et al. (2001). Briefly, the cDNA construct I-pRC/CMV was linearized with Not I and transcribed with Sp6 RNA-polymerase for the antisense riboprobe. The sense riboprobe was made by linearization with Xba I and transcription with T7 polymerase. Deparaffinized paraffin sections were rehydrated, treated with 5 μg/ml proteinase K (Roche Diagnostics; Mannheim, Germany), re-fixed with 4% paraformaldehyde /PBS, acetylated, and incubated with prehybridization buffer in a humid chamber at 51C for 2 hr. Hybridization was performed for 16 hr at 51C in the same buffer containing riboprobe (7.5 ng/ml). Specimens were washed twice with 2 × SSC containing 50% formamide and 1% 2-mercaptoethanol, followed by washing with 2 × SSC and 0.1 × SSC at 50C. The sections were blocked with 2% normal sheep serum and 0.05% Triton X-100 in 2 × SSC, washed twice with buffer 1 (50 mM Tris-HCl, pH 7.5, 225 mM NaCl), and incubated with alkaline phosphatase (AP)-conjugated anti-digoxygenin Fab (diluted 1:1000; Roche Diagnostics) in a humid chamber for 2 hr at 37C. Slides were washed twice with buffer 1 and equilibrated with 100 mM Tris-HCl, pH 9.5, containing 100 mM NaCl and 50 mM MgCl2. Endogenous AP was blocked with 5 mM levamisole. The digoxygenin-anti-digoxygenin conjugates were visualized by reaction with 0.15 mg/ml 5-bromo-4-chloro-3-indolyl phosphate/0.3 mg/ml nitroblue tetrazolium (FAST BCIP/NBT; Sigma) in 100 mM Tris-HCl, pH 9.5, containing 5 mM MgCl2, for 16 hr at 4C. Slides were then dehydrated and observed without counterstaining. Sections were hybridized with the sense and antisense probes in parallel and under the same conditions.
Digital Images
All digital images were captured at 512 × 512 pixels (for epifluorescence and brightfield microscopy) or 1024 × 1024 pixels (for EM and immunoEM) and processed in PhotoShop without changing the original appearance.
Immunoblotting of Tumors
Tumor tissue was homogenized with HEPES buffer (10 mM HEPES, 3 mM calcium acetate, pH 7.0; 1 ml buffer/100 mg tissue) without proteinase inhibitors in a glass-Teflon homogenizer. Aliquots of 1 ml homogenate were incubated at 37C for 0, 4, 8, 16, or 24 hr in the presence or absence of 0.6 U of human plasmin (Sigma) and centrifuged at 1000 rpm for 5 min to remove debris. Supernatants were resolved by 3-15% gradient SDS-PAGE gel and immunoblotted with perlecan antibodies R63, H5L5, A7L6, or 11B4 and appropriate HRP-conjugated secondary antibodies (swine anti-rabbit IgG, rabbit anti-mouse IgG, rabbit anti-rat IgG, all diluted 1:5000; DAKO). To detect rat perlecan with 11B4, homogenates were resolved by SDS-PAGE under nonreducing conditions.
Expression and Purification of Recombinant Perlecan
Perlecan construct I/II/III-pMT/V5-HisA was transfected into Drosophila S2 cells using a calcium phosphate transfection kit (Invitrogen). Briefly, 300 μl of DNA-CaCl2 solution containing 50 μg of I/II/III-pMT/V5-His A DNA and 2 μg of pCoHYGRO DNA was added dropwise to 300 μl of 2 ×HEPES-buffered saline with continuous mixing and incubated at RT for 30-40 min to form a fine calcium phosphate precipitate. The solution was then added dropwise to S2 cells (1 × 106 cells/ml) plated into T-25 flask 16 hr previously, and incubated with cells for 16-24 hr at 22-24C. Medium was removed, cells were washed twice, replated into the same flask in fresh complete medium, and incubated at 22-24C for 2 days, followed by hygromycin (300 μg/ml; Invitrogen) selection. As a control, 50 μg of empty vector and 2 μg of pCoHYGRO DNA were transfected into S2 cells. Antibiotic-resistant cells were treated with 1 mM CuSO4 in serum-free medium containing no hygromycin to induce perlecan expression. Immunoblotting of 20 μl of culture medium collected 3, 4 or 5 days after induction was performed to analyze perlecan expression.
To analyze GAG substitutions on recombinant perlecan, 1 ml of culture medium was collected 3 d after induction, lyophilized and resolubilized in heparinase buffer. Aliquots of 20 μl were treated with 1 milliunit of heparinase III (EC 4.2.2.8) and/or 5 milliunits of chondroitinase ABC (EC 4.2.2.4; Seikagaku America) as described previously. Samples were resolved by 7% SDS-PAGE gel and immunoblotted with biotin-conjugated stub antibodies F69-3G10, 1-B-5, 2-B-6, or 3-B-3. R63 was used to probe perlecan core protein. HRP-conjugated streptavidin (Amersham Biosciences) was used to detect stub antibodies at 1:1500.
To purify recombinant perlecan, S2 cells (5 × 106 cells/ml) were maintained in 500 ml of serum-free medium (without hygromycin) for 3 days at 22-24C followed by 1 mM CuSO4 treatment for another 3 days. S2 Cells were pelleted at 1000 × g for 2-3 min. The supernatant was applied on a column of DEAE-Sepharose 4 fast flow (2.5 × 5 cm; Amersham Biosciences) equilibrated in 50 mM Tris-HCl, pH 8.0, containing 0.2 M NaCl, 4 M urea, 10 mM N-ethylmaleimide, 20 mM EDTA, 0.2 mM phenylmethylsulfonyl fluoride, and 0.1% Tween-20. After loading, the column was washed extensively in equilibration buffer, then with a buffer in which the Tris-HCl was replaced with 50 mM sodium acetate, pH 4.0. The column was then sequentially washed with 0.2 M sodium acetate, pH 5.0, 1.1 M sodium acetate, pH 5.0, and 0.2 M sodium acetate, pH 7.0, before elution with 2.0 M sodium acetate, pH 7.0. The peak fractions were pooled and concentrated by Ultrafree-15 centrifugal filtration (Millipore; Billerica, MA), replacing elution buffer with PBS during concentration. The purity of perlecan was analyzed by silver staining of 3-15% SDSPAGE gels.
Results
Expression of HSPGs by RT101 Cells
Perlecan, agrin and type XVIII collagen are HSPGs present in BMs (reviewed in Dunlevy and Hassell 2000). Our laboratory has reported that RT101 cells synthesize perlecan (Tapanadechopone et al. 2001). To identify whether agrin and type XVIII collagen were synthesized by RT101 cells, PGs were prepared from serum-free medium conditioned by RT101 cells and subjected to heparinase III and chondroitinase ABC treatment. As shown in Figure 1, RT101 cells can synthesize agrin and type XVIII collagen in addition to perlecan. Perlecan (Figure 1A) was an HSPG as reported previously; two core proteins polypeptides were consistently detected. Agrin (Figure 1B) contained not only HS chains but perhaps also CS chains because combined heparinase III and chondroitinase ABC treatment gave rise to a sharper resolution of the core protein of slightly smaller mass (~200 kD; Lane HC) compared with heparinase III treatment alone (Lane H). Additional bands were detected in Lanes HC and C compared with Lanes U and H. These bands might be degraded products of agrin, which carried CS chains before chondroitinase ABC treatment. Type XVIII collagen (Figure 1C) was synthesized as an HSPG. The largest diffuse material (>300 kD) was an incompletely digested product; the lower band indicated by the arrow represented the core protein of type XVIII collagen (~180 kD). The smaller polypeptides might be degraded products. In Figure 1D, the blot was probed with antibody against HS stub chains. Bands corresponding to perlecan (P), agrin (A), and type XVIII collagen (C) were detected. Because a monoclonal antibody was used to detect perlecan, whereas polyclonal antibodies were used for the other PGs, the blots are not indicative of relative amounts. Perlecan was the most abundant HSPG in RT101 medium and was the focus of further work. However, the effects of agrin and type XVIII collagen on tumor growth are discussed below.
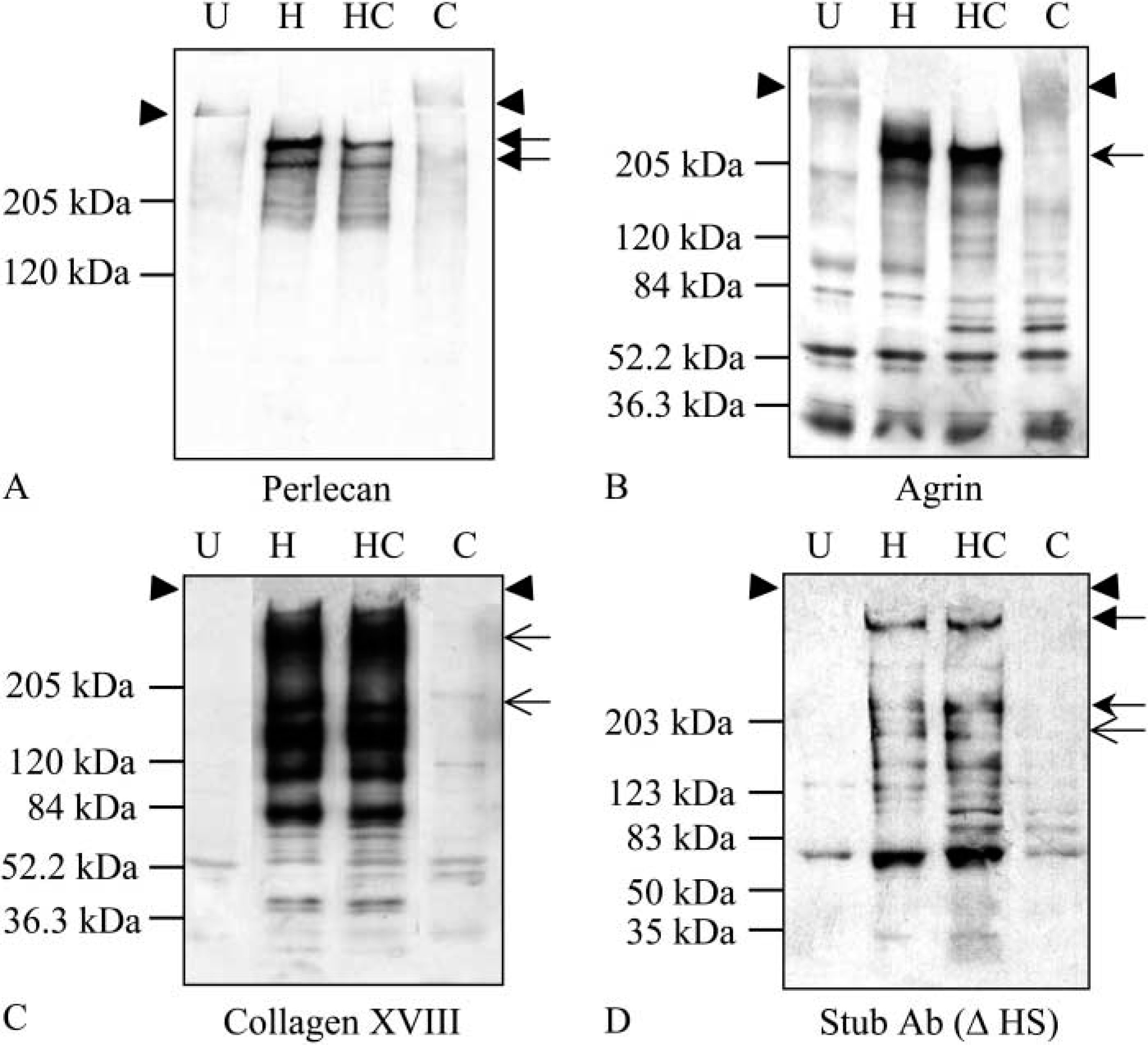
Expression of BM HSPGs by RT101 cells. Serum-free medium conditioned by RT101 cells for 24 hr was untreated (U) or treated with heparinase III (H), chondroitinase (C), or heparinase III and chondroitinase (HC) at 37C overnight. The samples were resolved by 3-15% SDS-PAGE, followed by membrane transfer and Western blotting. (
Tumor Morphology and Immunostaining of Tumor Sections
RT101 cells grown in vivo showed large nuclei with prominent nucleoli (Figures 2A-2C and 2E), which is typical for tumor cells. Large blood vessels (Figure 2C) and capillaries (Figures 2C, 2D, and 2F) were observed on tumor sections, indicating massive vascularization within the tumors. The BMs surrounding capillary endothelial cells appeared to be thick (Figures 2C, 2D, and 2F).
Both tumor and host cells can synthesize perlecan. To identify the source(s) of perlecan in mouse epidermal tumors generated in immunocompromised rats, immunofluorescence staining of tumor frozen sections with species-specific mouse anti-rat and rat anti-mouse perlecan antibodies was performed. The results showed that perlecan deposited around blood vessels was of both host and tumor cell origin, and tumor-derived perlecan was also widely distributed in tumor matrix (Figures 3A-3C). The co-localization of tumor and host perlecan in the BMs of blood vessels was further shown by immunoEM, which revealed that 10-nm gold-labeled tumor perlecan and 15-nm gold-labeled rat perlecan co-distributed in the BMs (Figures 4A and 4B). Controls confirmed the staining specificity (Figures 4C-4E). To exclude the possibility that RT101 tumor cells were contaminated with endothelial cells, which would result in positive staining of blood vessels, both RT101 cells in culture and tumor sections were stained for the endothelial cell surface marker PECAM-1. A lack of labeling of tumor sections with anti-mouse PECAM-1, but labeling with anti-rat PECAM-1, confirmed the host origin of blood vessels in tumor sections (Figures 3D-3F). RT101 cells in culture were also negative for mouse PECAM-1 (data not shown), indicating that mouse perlecan present in rat blood vessels was synthesized by mouse epidermal tumor cells only. Because perlecan can interact with other BM components, including type IV collagen, laminin, and entactin/nidogen-1, through its HS chains or core protein (Battaglia et al. 1992; Ettner et al. 1998; Hopf et al. 2001), the distributions of these molecules in the tumors were also analyzed. As shown in Figures 3G-3I, type IV collagen was deposited throughout the tumor matrix and around blood vessels. Laminin γl-chain derived from host cells was distributed around blood vessels only (Figure 3N), while laminin-5 synthesized by tumor cells was deposited pericellularly (Figures 3J-3L). Interestingly, entactin/nidogen-1 derived from tumor cells was distributed only around blood vessels (Figure 3M). The absence of tumor-derived entactin/nidogen-1 in the tumor matrix was unexpected. Because of the unavailability of monoclonal antibodies to differentiate mouse and rat type IV collagen as well as mouse laminin and rat entactin/nidogen-1, we were unable to determine whether these perlecan-binding BM components also had a dual source in tumor vascular BMs. Species-specific antibodies against agrin and type XVIII collagen were also unavailable.
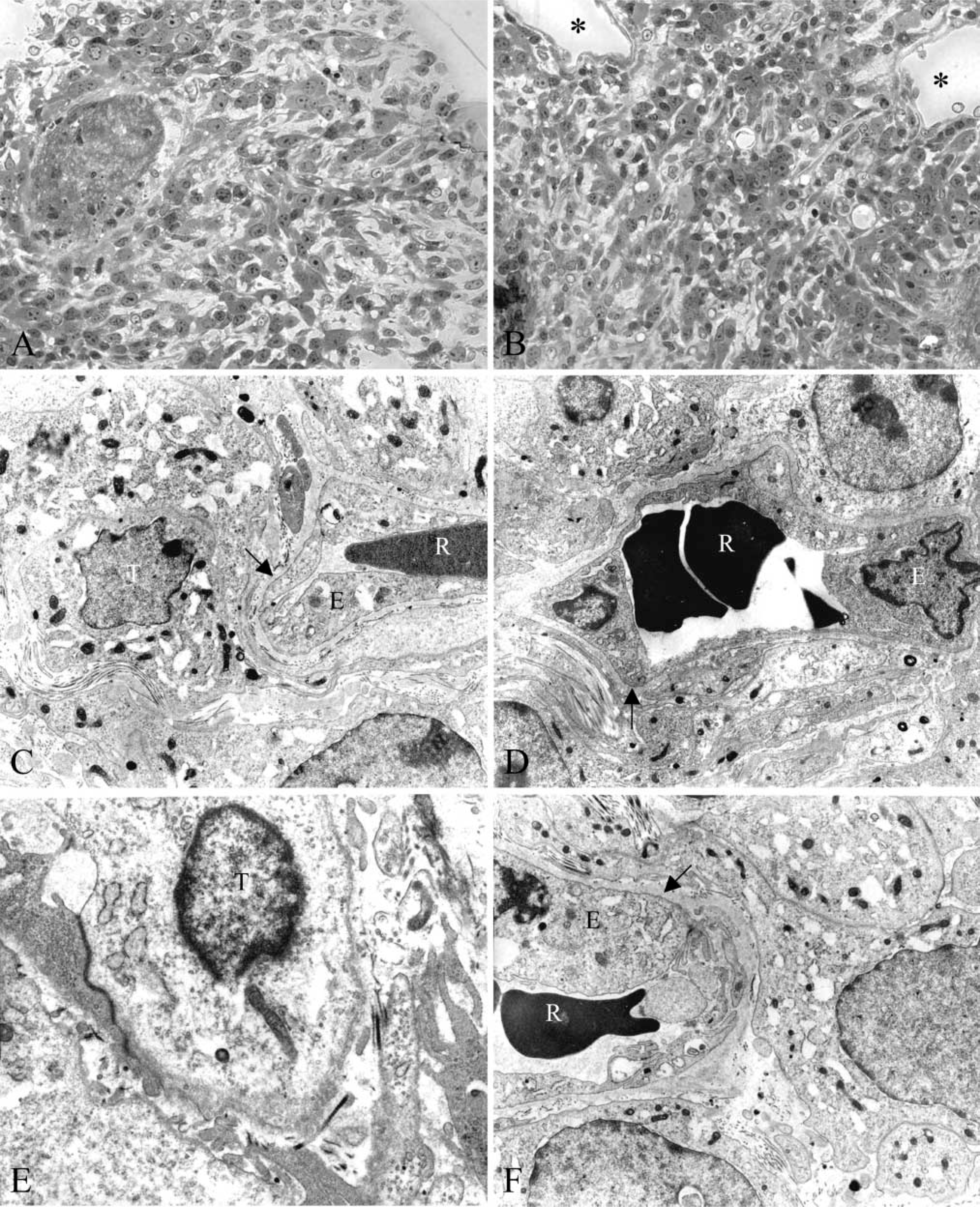
Morphology of tumor. (
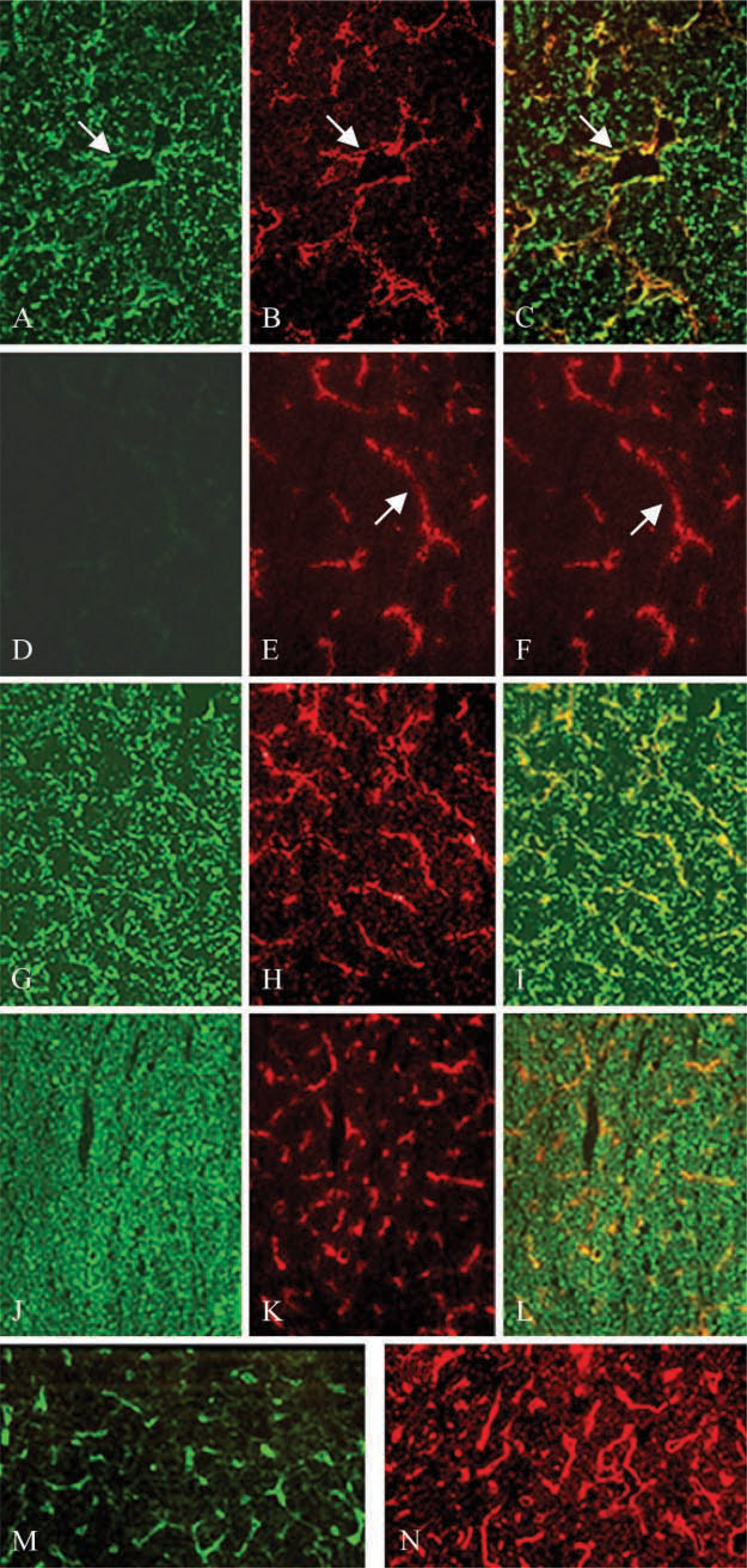
Immunofluorescence staining of tumors. Tumor frozen sections were stained with monoclonal rat anti-mouse perlecan antibody G9L1 (
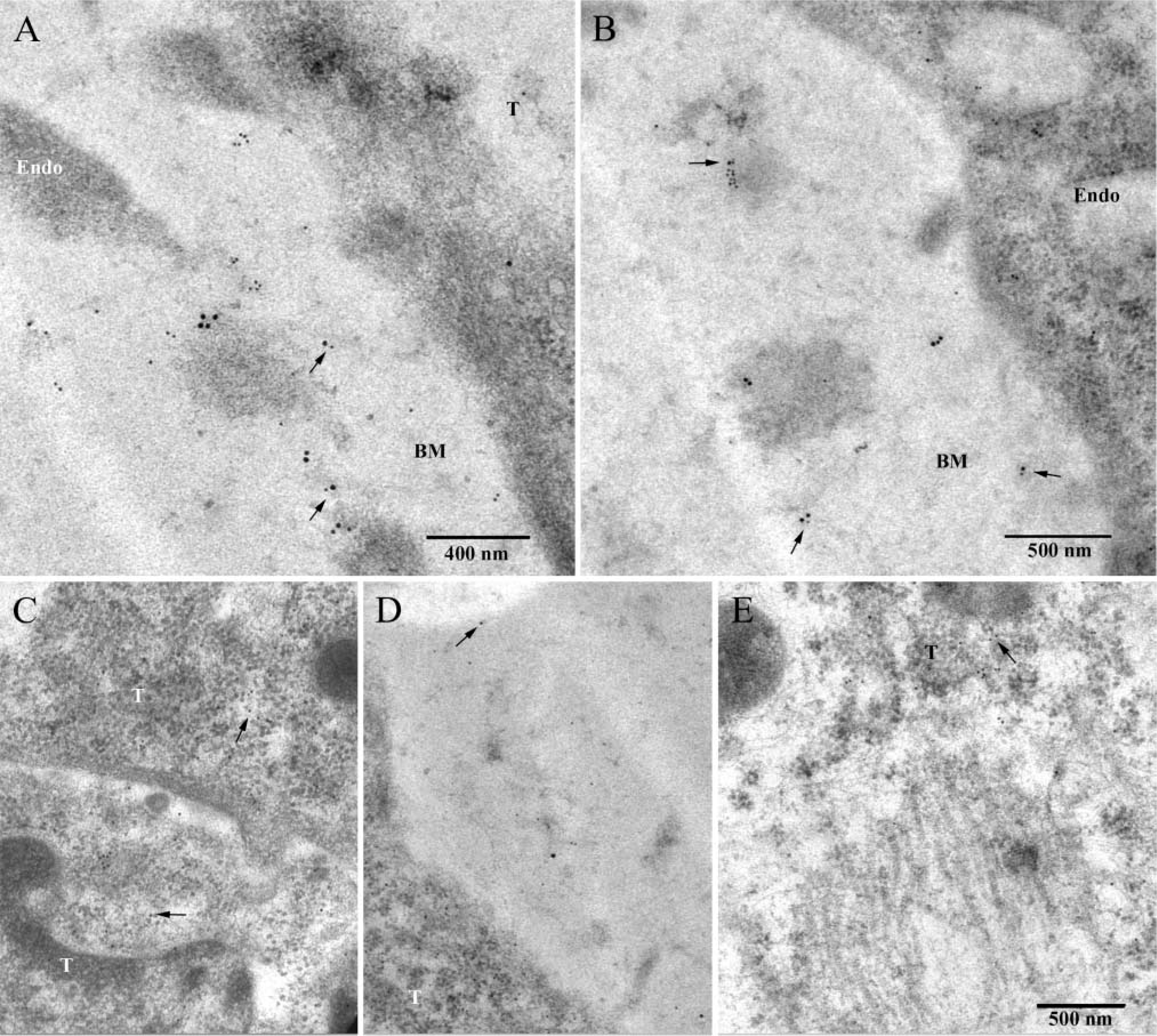
Immunoelectron microscopy. (
Matrix Incorporation of Tumor and Host Perlecan
Perlecan was integrated into the tumor ECM and host BMs. To assess whether host and tumor perlecan were differentially extractable, tumor tissue was homogenized with 10 mM HEPES buffer and incubated at 37C for up to 24 hr. Soluble mouse perlecan was detected by antibodies against domain III (H5L5) or domain IV (A7L6) 4 hr after incubation, even without EDTA or protease treatments, and no significant degradation was observed in the absence of protease inhibitors as long as the incubation was less than 16 hr (not shown). Compared with mouse perlecan, rat perlecan was more stable. Only small amounts of rat perlecan were released until 8-hr incubation in the presence of plasmin (not shown). The results suggest that rat perlecan is integrated into vascular BMs more tightly than matrix tumor perlecan and that protease(s) such as plasmin are required to interrupt the interactions between rat perlecan and other BM components.
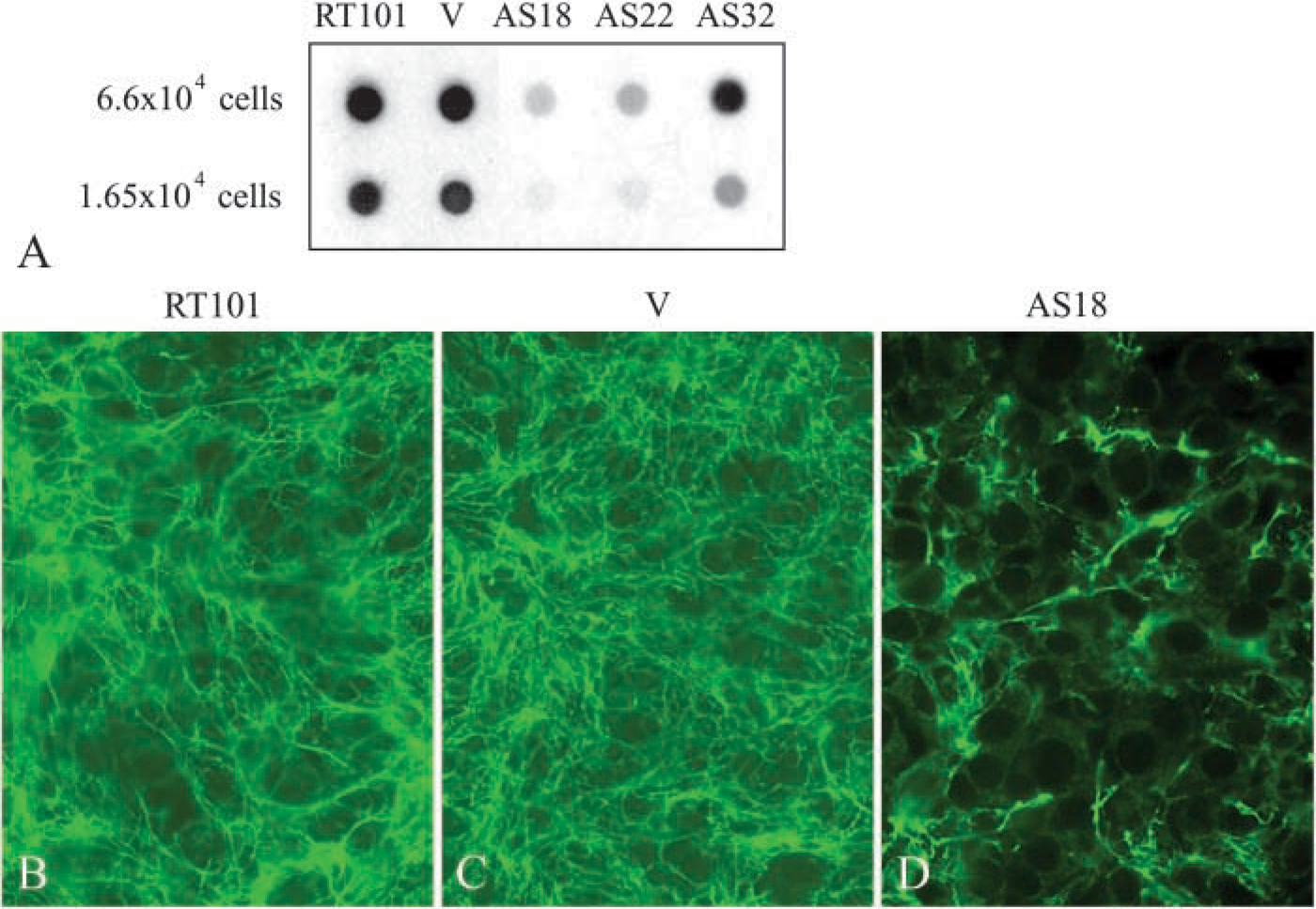
Analysis of perlecan level in antisense perlecan-transfected cells. (
Generation of Antisense-perlecan Clones
The antisense perlecan-targeted cells were expanded by ring cloning. As shown in Figure 5A, antisense perlecan clones 18 (AS18) and AS22 produced much lower levels of perlecan compared with untransfected or vector-transfected cells, and AS32 had an intermediate perlecan level compared with control cells, AS18 and AS22. The expression level of perlecan in AS18 was further analyzed by immunofluorescence staining. The results showed that untransfected and vector-transfected cells deposited a large amount of perlecan into the ECM 4 days after plating (Figures 5B and 5C), whereas AS 18 had significantly lower levels of perlecan in the ECM (Figure 5D).
Expression and Purification of Recombinant Perlecan
The expression of perlecan in S2 cells was examined by immunoblotting 3, 4, and 5 days after induction. High expression of perlecan was seen 3 days after induction (data not shown). A representative result of the purification of perlecan is shown in Figure 6A. The yield of perlecan was ~1 mg/liter. Silver staining showed the high purity of perlecan (Figure 6B, Lane 2). Western blotting (Figure 6B, Lane 1) suggested that no degradation of perlecan occurred during the process of purification. GAG analysis revealed that perlecan synthesized by S2 cells contained only HS chains (Figure 6C). Although anti-HS stub antibody detected a single species after heparinase III ± chondroitinase ABC treatment, CS or DS chains were not detected by stub antibodies against CS/DS after chondroitinase ABC treatment (data not shown). The small increase in perlecan core protein migration after heparinase III treatment suggests that the HS chains synthesized by S2 cells are very small (Figure 6D).
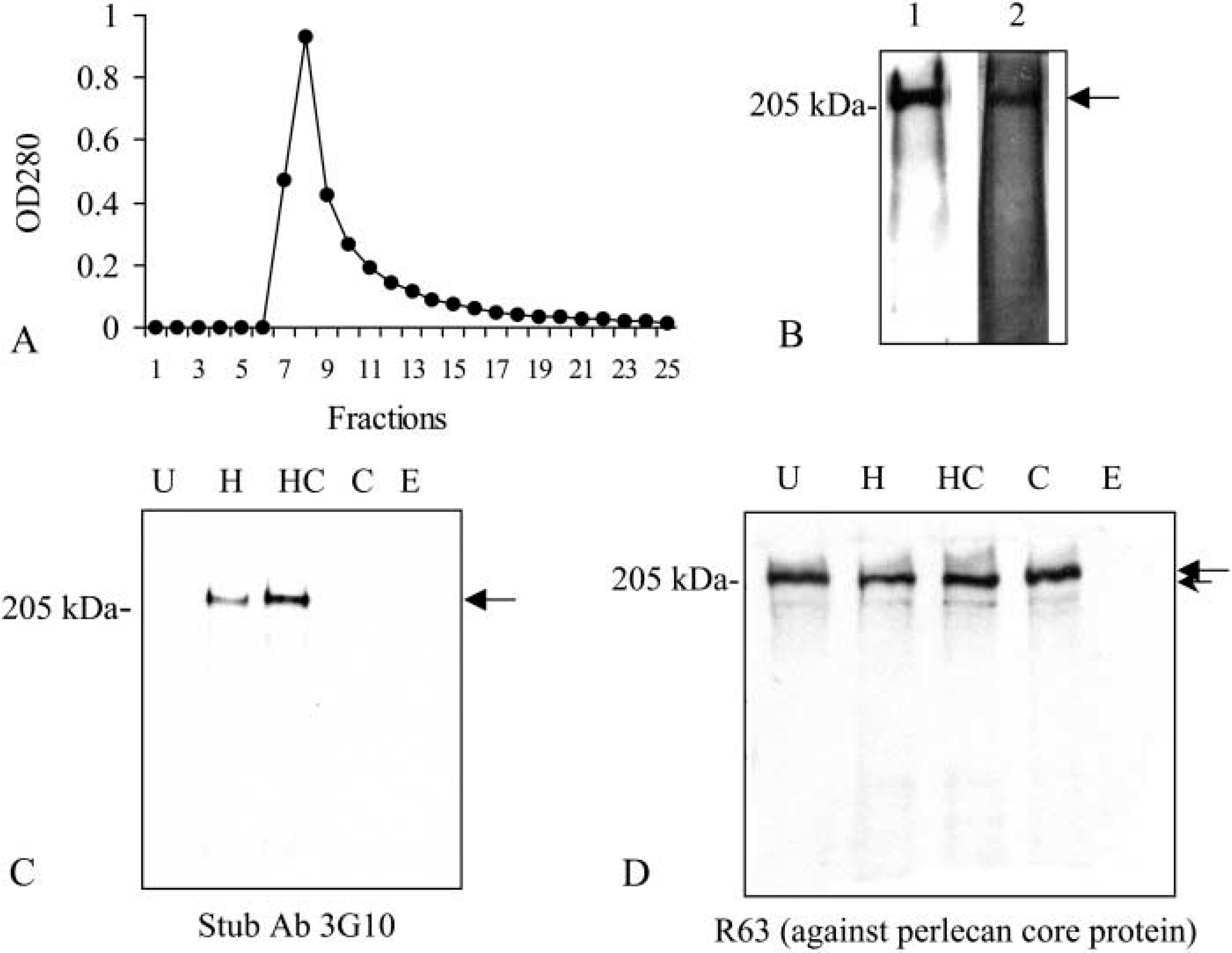
Purification and characterization of recombinant perlecan expressed in Drosophila S2 cells. (
Growth of Antisense Perlecan Clones In Vitro
The growth rates of untransfected and transfected cells were examined by cell counting. As shown in Figure 7B, untransfected and vector-transfected cells had similar high growth rates. AS18 and AS22 cells grew much more slowly than control cells, and AS32 cells had an intermediate growth rate. The doubling times of untransfected, vector-transfected, AS18, AS22, and AS32 were 15.31 ± 1.11, 14.24 ± 0.72, 28.65 ± 0.71, 21.99 ± 0.21, and 18.37 ± 3.11 hr, respectively. The growth rates and cell doubling times corresponded to the perlecan expression levels in control and antisense perlecan cells (Figure 5A), indicating that perlecan is correlated with the growth rate. To confirm this observation, perlecan was added to cultures of antisense perlecan and vector-transfected cells for 3 days. Recombinant perlecan (10 μg/ml) increased the growth of AS18 cells by 126% (Figure 7A), whereas it had no significant effects on the growth of vector-transfected cells.
HS chains on perlecan can bind and modulate the activity of heparin-binding growth factors such as FGF-2 (Aviezer et al. 1994). To examine whether the absence of HS chains of perlecan was responsible for cell growth retardation, 1 × 104 cells were plated into 24-well plates and cultured in the presence of heparin for 3 or 4 days. The results showed that heparin had no effects on the growth of untransfected, vector-transfected, AS18, and AS22 cells (Figures 7A and 8A), suggesting that perlecan regulates cell growth through its core protein rather than its HS chains.
RT101 cells express FGF-2 and FGF-7 at both the mRNA and the protein levels (Jiang et al. unpublished data). Untransfected RT101 and AS22 cells grown in the presence of FGF-2 showed increased cell growth, whereas FGF-7 had no effects (Figure 8A), indicating that FGF-7 is not mitogenic for RT101 cells. The biological function of FGF-7 is specifically mediated through FGFR-2 isoform IIIb, which is normally expressed on epithelial cells (reviewed in Powers et al. 2000). RT-PCR analysis of FGFR-2 expression showed that RT101 cells had an absence of IIIb isoform, whereas the IIIc isoform, which is normally expressed on mesenchymal cells, was detected (Figure 8B). These data are consistent with the lack of responsiveness to FGF-7 but sensitivity to FGF-2. The switched expression of FGFR-2 isoforms in transformed epithelial cells was consistent with previous reports (Carstens et al. 1997; Feng et al. 1997).
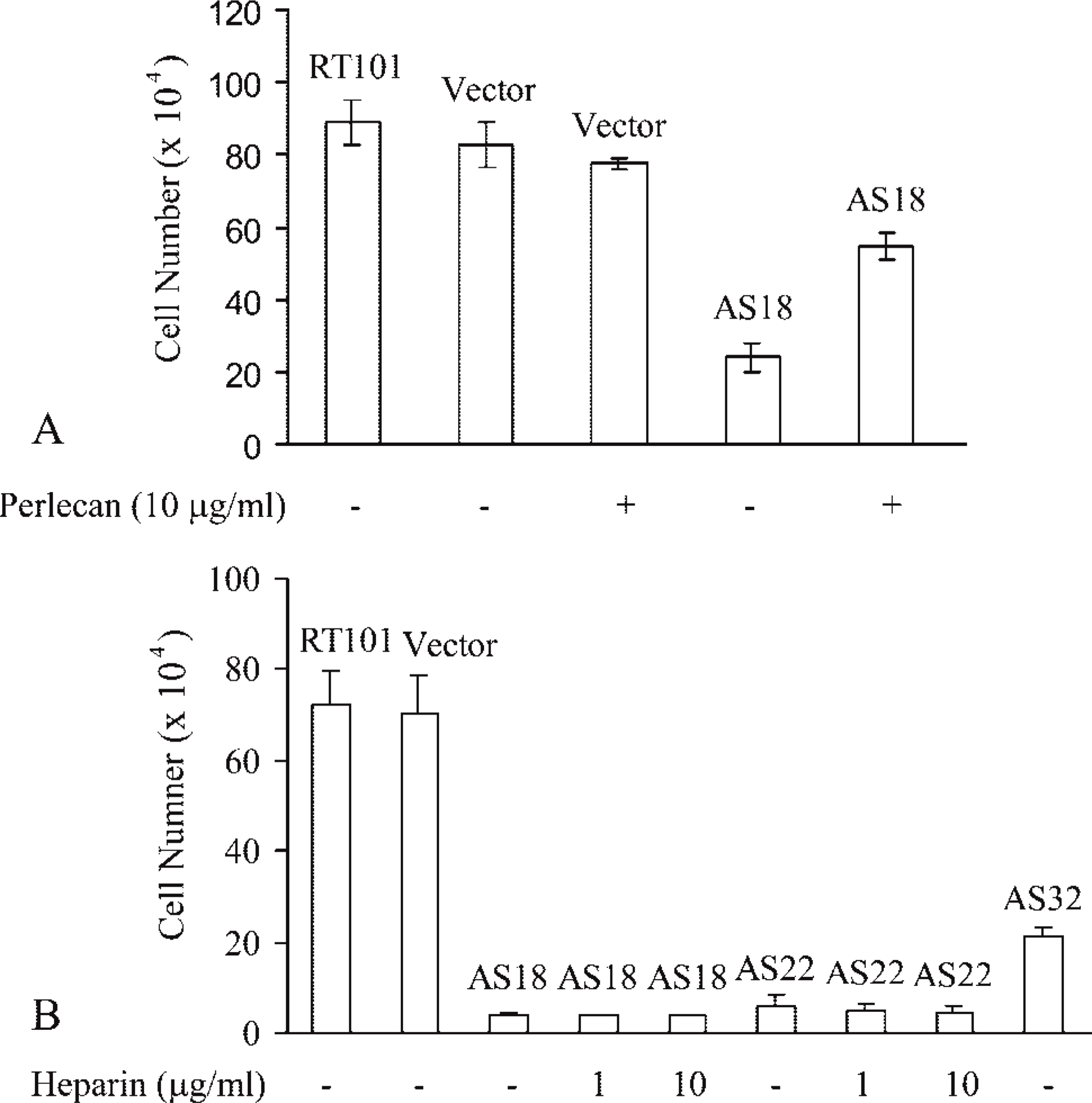
The growth of antisense-perlecan clones in vitro. (
Growth of Antisense Perlecan Clone In Vivo
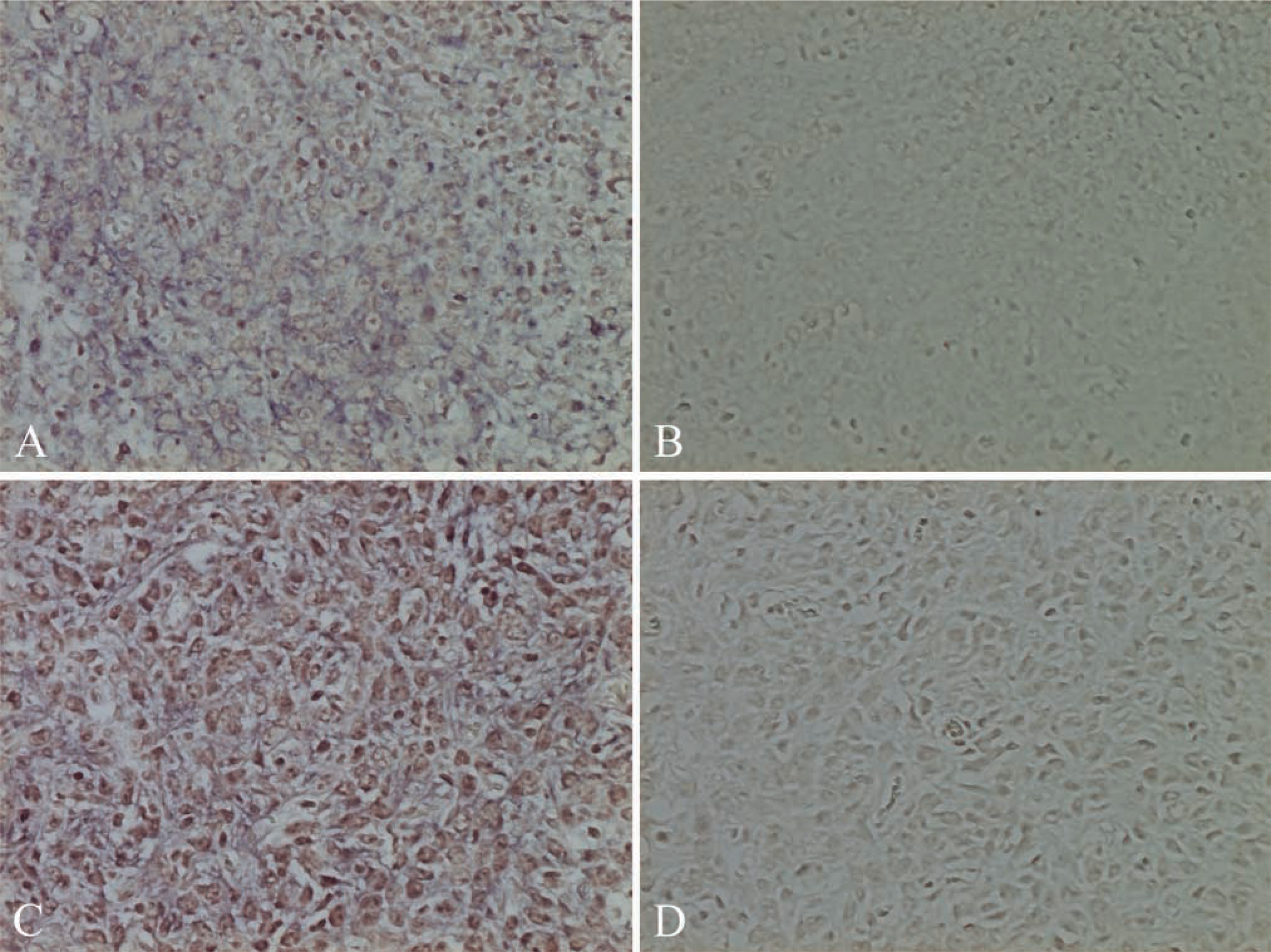
To examine whether the reduced perlecan level would inhibit tumor growth in vivo, control cells or AS18 cells (1 × 106) were injected intradermally into immunocompromised rats. AS18 cells induced no tumors in vivo (n=5), whereas three tumors (1.73 ± 2.01 g) were produced by untransfected cells on the contralateral sides. Prominent tumors were also produced by vector-transfected cells (61.78 ± 26.23 g; n=3). ISH (Figure 9) and PECAM-1 staining (Figure 10) showed high perlecan mRNA levels and massive neovascularization in tumors generated by untransfected and vector-transfected cells. These data suggest that a reduced perlecan level can inhibit tumor growth completely in our tumor model.
Discussion
Tumor growth and neovascularization are accompanied by a pronounced increase in perlecan expression (Guelstein et al. 1993; Cohen et al. 1994; reviewed in Iozzo et al. 1994; Iozzo 1998; Roskams et al. 1998). In this study we used species-specific perlecan monoclonal antibodies, which were generated and characterized in our laboratory (Couchman and Ljubimov 1989; Couchman et al. 1995, 1996), to analyze the source(s) of perlecan in mouse epidermal tumors produced in immunocompromised rats. Both immunofluorescence staining and immunoEM showed that perlecan deposited around blood vessels was of both host and tumor cell origin. In addition, tumor perlecan was also distributed throughout the tumor matrix. To our knowledge, this is the first report of the co-localization of host and tumor perlecan in tumor vascular BMs. The distribution pattern of tumor perlecan suggested that it was diffusible, while the absence of host perlecan in the tumor matrix indicated that host perlecan was tightly integrated into the blood vessel walls. Antisense perlecan-expressing cells produced no tumors in vivo although host perlecan was not manipulated, suggesting that tumor perlecan but not host perlecan is essential for tumor growth and angiogenesis in our tumor model.
Western blot analysis of tumor and host perlecan indicated that tumor perlecan was more easily solubilized than host perlecan and did not require plasmin treatment for release. This, together with the distribution patterns of tumor and host perlecan in tumor sections, suggests that tumor and host perlecan differ in diffusion ability. This might explain the different contributions of tumor and host perlecan to tumor growth and angiogenesis. In the process of tumor growth, tumor perlecan, probably mobilized by proteases (White-lock et al. 1996), diffuses in the tumor matrix, recruits tumor-derived angiogenic growth factors, possibly FGF-2 and FGF-7, through the core protein and/or HS chains, and then is incorporated into newly forming host blood vessels. The incorporation of tumor perlecan into host blood vessels brings highly concentrated angiogenic growth factors to the vicinity of blood vessels, resulting in massive neovascularization, which further stimulates tumor growth. Host perlecan, which is tightly integrated into blood vessel BMs, appears to be more stable, does not diffuse to or appear in the tumor matrix. In the absence of tumor perlecan or when the tumor perlecan level is low, host perlecan may be unable to recruit sufficient angiogenic growth factors to the vicinity of blood vessels, resulting in limited tumor growth and angiogenesis.
Responsiveness of untransfected and antisense-perlecan cells to FGF-2 and −7. (
ISH of tumor paraffin sections. Tumors generated by untransfected (
The mechanism of the incorporation of tumor perlecan into host blood vessels remains to be studied. Tumor perlecan may interact with host perlecan through domain V (Yurchenco et al. 1987) and/or may interact with other BM components, such as laminin, type IV collagen (Battaglia et al. 1992; Ettner et al. 1998), entactin/nidogen-1, −2, and fibulin-2 (Hopf et al. 2001), through its HS chains and/or core protein. Multiple intermolecular interactions may facilitate the incorporation of perlecan into vascular BMs. It would be interesting to know whether tumor perlecan incorporated into the vascular BMs can exchange with that still in the tumor matrix. If tumor perlecan in the vascular BMs is exchangeable, then the intermolecular interactions between tumor perlecan and BM components will differ from those of host perlecan with other BM components. Given that the core protein of perlecan is conserved from C. elegans and Drosophila to mouse and human, whereas the HS chains on perlecan are highly variable (Hassell et al. 1980; Mohan and Spiro 1991; Molist et al. 1998; Tapanadechopone et al. 2001), HS chains might be the major factor that dictates the intermolecular interactions between perlecan and other BM components. The different substructure of HS chains might influence not only the diffusion ability but also the growth factor-binding ability of perlecan.
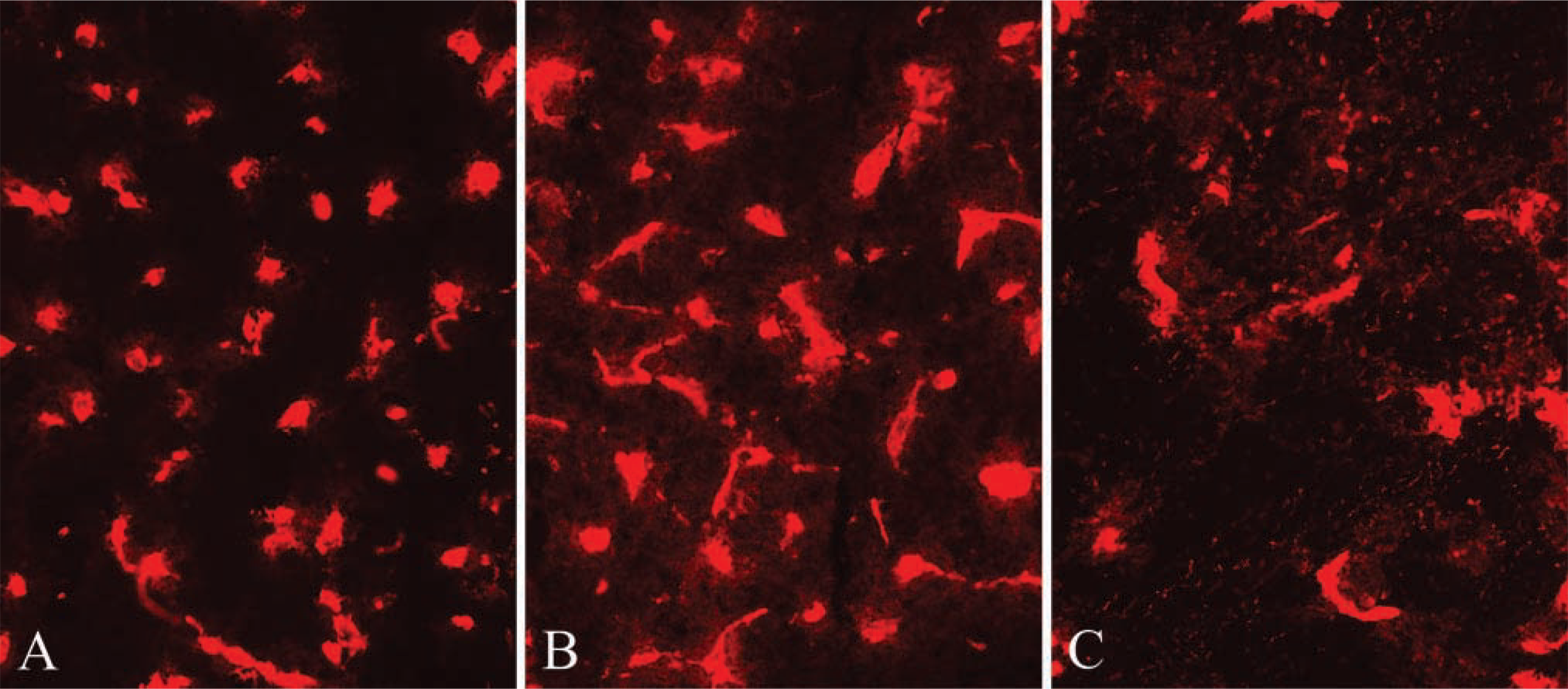
Immunofluorescence staining of tumor frozen-sections with PECAM-1 antibody TLD-3A12. Extensive angiogenesis was observed in tumors generated by untransfected (
Among the other major BM components, type IV collagen was distributed in tumor matrix and blood vessels, similar to the distribution pattern of tumor perlecan. However, because of the unavailability of species-specific antibodies against mouse and rat type IV collagen, it is unclear whether type IV collagen also had a dual source in tumor vascular BMs. Tumor-derived laminin-5 had a pericellular distribution. Tumor-derived entactin/nidogen-1 had the same distribution pattern as host laminin; both were present in blood vessels and no matrix distribution was observed. Entactin/nidogen-1 can interact with both perlecan (Hopf et al. 2001) and laminin (Paulsson et al. 1987). The absence of tumor entactin/nidogen-1 in tumor matrix suggests that tumor entactin/nidogen-1, which is somehow diffusible in the tumor matrix, has a much higher affinity for host-derived laminin than that for tumor-derived perlecan. Laminin molecules are cross-shaped heterotrimers composed of α-, β-, and γ-chains. Five α-chains, three β-chains, and three γ-chains have been identified, and 12 distinct laminin isoforms have been isolated thus far. All laminin isoforms, except for laminin-5 (α3β3γ2) and laminin-12 (α2β1γ3), share the γ1-chain (reviewed in Colognato and Yurchenco 2000). The interaction between entactin/nidogen-1 and laminin is laminin isoform-specific. A single LE (laminin EGF-like) module, γ1III4, has been identified as exclusively responsible for the binding of entactin/nidogen-1. The laminin γ2- chain is inactive because of two crucial substitutions in the entactin/nidogen-binding epitope (reviewed in Mayer et al. 1998). Therefore, it is not surprising that tumor-derived laminin-5 cannot retain tumor-derived entactin/nidogen in the tumor matrix. Tumor-derived entactin/nidogen-1 might be driven to the blood vessel walls through intermolecular interactions with host laminin. The significance of the blood vessel localization of tumor entactin/nidogen remains to be studied; it might contribute to the angiogenic process.
Perlecan is a major storage site for growth factors in ECM and BMs. The HS moieties of perlecan can bind a variety of heparin-binding growth factors, including FGF-1, −2, −4, −7, heparin-binding epidermal growth factor (HB-EGF), and transforming growth factor (TGF)-β (Aviezer et al. 1994; reviewed in Iozzo and San Antonio 2001; Tapanadechopone et al. 2001), indicating that HS chains of perlecan play important roles in regulating cell behavior. However, several lines of evidence suggest that perlecan core protein maybe a potent growth regulator in our system. First, characterization of agrin and type XVIII collagen showed that both contain HS chains. Second, cell surface HSPGs, including syndecan-1, −2, and −4, are expressed on RT101 cells (unpublished data). Third, the growth rate of antisense perlecan cells was not affected by the presence of exogenous heparin, indicating that a lack of HS chains could not explain a retardation of cell growth in the absence of perlecan. Fourth, recombinant perlecan (domains I, II, and III but lacking IV and V) could restore proliferation of AS18 cells, and this perlecan from Drosophila S2 cells has only very small HS chains.
Both FGF-2 and −7 are synthesized by RT101 cells. Moreover, FGF-7 can interact with perlecan core protein and heparan sulfate (Ghiselli et al. 2001; Tapana-dechopone et al. 2001). Correlating with the lack of FGF-7 receptor (FGFR-2IIIb), RT101 cells were not responsive to FGF-7. The wild-type RT101 and AS22 cells were responsive to FGF-2, although FGF-2 requires HS chains for activity. The fact that AS22 cells could respond to FGF-2 implies that a cell-surface HS other than that of perlecan must be functional. Furthermore, the effects of perlecan core protein on proliferation must, in this case, be independent of FGF-7. As has been noted before in transformed epithelial cells (Carstens et al. 1997; Feng et al. 1997), RT101 express the FGFR-2IIIc isoform, consistent with FGF-2 mediated mitogenesis. FGF-2 has not been reported to interact with perlecan core protein, and it is therefore likely that factors other than HS chains FGF-2 and −7 may regulate perlecan-mediated cell growth in our system.
Collectively, tumor perlecan was both mitogenic and angiogenic in our tumor model, and targeting perlecan might be an efficient way to treat solid tumors clinically.
Footnotes
Acknowledgements
Supported in part by National Institutes of Health Grant AR-36457 to JRC, the Deutsche Forschungsgemeinschft (SFB 492, project B10) and by the Interdisciplinary Center for Clinical Research, University of Muenster (Project D18) to LS and RS.
We thank Dr John R. Hassell for providing us with the perlecan constructs I/II/III-pBS and I/II/III-pRC/CMV, Dr Jonathan R. Jones for the laminin-5 antibody J18, and Dr Gregory J. Cole for the agrin antibody. We thank the Developmental Studies Hybridoma Bank for providing us with laminin monoclonal antibody D18. We thank Daniel Mihalik and Andrea Babulova for their kind help in tumor harvesting and sample preparations. We thank Dr Anne Woods for her helpful advice throughout the study.