
Abstract
Tumor markers are widely used in pathology not only for diagnostic purposes but also to assess the prognosis and to predict the treatment of the tumor. Because tumor marker levels may change over time, it is important to get a better understanding of the molecular changes during tumor progression. Occurrence of breast and ovarian cancer is high in older women. Common known risk factors of developing these cancers in addition to age are not having children or having children at a later age, the use of hormone replacement therapy, and mutations in certain genes. In addition, women with a history of breast cancer may also develop ovarian cancer. Here, the authors review the different tumor markers of breast and ovarian carcinoma and discuss the expression, mutations, and possible roles of cell surface heparan sulfate proteoglycans during tumorigenesis of these carcinomas. The focus is on two groups of proteoglycans, the transmembrane syndecans and the lipid-anchored glypicans. Both families of proteoglycans have been implicated in cellular responses to growth factors and morphogens, including many now associated with tumor progression.
Breast Cancer
Background
Breast cancer is the most frequently diagnosed cancer in women; one in four new cancers diagnosed worldwide each year is a cancer of the female breast. Breast cancer is also the leading cause of cancer-related death in women worldwide (Kamangar et al. 2006; Jemal et al. 2009; La et al. 2010). Although multiple risk factors, such as old age, family history, and hormonal factors, have been identified, the etiology of the disease remains incompletely understood (Dumitrescu and Shields 2005).
Carcinoma is the commonest malignant tumor of the breast. Ductal carcinoma in situ (DCIS) and invasive carcinoma are differentiated. Breast carcinomas are derived from the epithelial cells that line the terminal duct lobular unit. Cancer cells that remain within the basement membrane of the elements of the terminal duct lobular unit and the draining duct are classified as in situ or noninvasive. An invasive breast cancer is one in which there is dissemination of cancer cells outside the basement membrane of the ducts and lobules into the surrounding adjacent normal tissue. The most commonly used classification of invasive breast cancers divides them into ductal and lobular types. This classification was based on the belief that ductal carcinomas arose from ducts and lobular carcinomas from lobules. We now know that invasive ductal and lobular breast cancers both arise from the terminal duct lobular unit, and this terminology is no longer appropriate (Sainsbury et al. 2000).
Breast cancer is not a single disease but is highly heterogeneous at both the molecular and clinical levels (Perou et al. 2000; Sorlie et al. 2001; Bergamaschi et al. 2006; Chin et al. 2006; Vargo-Gogola and Rosen 2007). As a result, the dilemma facing clinicians who treat breast cancer patients has been for many years how to avoid overtreatment and undertreatment. Markers such as estrogen receptor (ER), progesterone receptor (PR), and epidermal growth factor receptor 2 (HER2) are currently used in the clinic for prognostic purposes as well as to stratify patients for appropriately targeted therapies (Harvey et al. 1999; Subramaniam and Isaacs 2005; Ross et al. 2005; Payne et al. 2008). Estimates suggest that treatment failure presently occurs in approximately 30% of breast cancer patients (Mullan and Millikan 2007).
Histopathology
Although several histologic types and subtypes of mammary carcinoma exist, >95% are either ductal or lobular carcinoma (Tavassoli 1999; Rosen PP 2001). Both of these types of mammary carcinoma can be in situ and/or infiltrating, the difference being whether or not the neoplastic cells have invaded through the outer myoepithelial cells and into the surrounding intra- or interlobular stroma (Rosen PP 2001). The majority (75%–80%) of mammary carcinomas are ductal carcinomas, whereas classic infiltrating lobular carcinoma (ILC) comprises 10% to 15% of all mammary carcinomas. Overall, ILC has a better prognosis compared with ductal carcinoma. The incidence of ILC has been steadily increasing over the past 20 years. Although the precise cause of this remains unknown, data support that it might be related to the use of hormone replacement therapy among postmenopausal women (Li et al. 2003).
ER, PR, and HER2 status are three important molecular features of invasive breast carcinomas that have been identified during the past 30 years, and their evaluation by pathologists is now mandatory (Allred 2010). ER and PR are growth-regulating nuclear transcription factors that are usually measured by immunohistochemistry (IHC), and the amount of protein expressed is directly related to responsiveness to endocrine therapy, which is why they are so important (Harvey et al. 1999; Mohsin et al. 2004). HER2 is a growth factor receptor (among other functions) at the cell membrane. Cell surface levels of this membrane protein strongly associate with amplification of this oncogene, and thus HER2 status can be measured either at the protein level by IHC or at the DNA level by assessing gene copy number with assays such as fluorescence in situ hybridization. Overexpressed and/or amplified HER2 is a relatively weak prognostic factor in untreated patients, but it is a strong predictive factor for responsiveness to targeted therapies such as trastuzumab, which is the primary reason for measuring it (Ross et al. 2004; Allred 2010).
Ovarian Cancer
Epidemiology and Risk Factors
Epithelial ovarian cancer is the leading cause of death among the gynecologic cancers and the fourth leading cause of cancer death in women in the United States (Jemal et al. 2009). Incidence rates vary considerably but are highest in industrialized countries with the exception of Japan. Ovarian cancer is one-tenth as common as breast cancer but three times as lethal. The high mortality rate is generally attributed to its occult development, resulting in advanced, widespread disease occurring in approximately 75% of women at diagnosis. The overall 5-year survival rate of ovarian cancer is about 30%. However, in about 25% of women, disease is confined to the ovary (stage I), and 5-year survival is more than 90%, compared with 10% for women with advanced disease (Christie and Oehler 2006; Kurman et al. 2008). The incidence of epithelial ovarian cancer is age related and is generally a disease of postmenopausal women. Malignant epithelial tumors are most common between the ages of 40 and 60 years.
The molecular events leading to the development of epithelial ovarian cancer are not known currently. Epidemio-logically developed risk factors that have been identified include family history, null parity, early menarche and late menopause, increasing age, and residence in developed countries (Daly and Obrams 1998). Most ovarian cancers are the result of sporadic mutations. Approximately 10% of cases are attributed to a familial disposition. Women who carry deleterious mutations of the BRCA1 or BRCA2 gene are the largest subset at risk for ovarian cancer. For women who have BRCA1 (17q21chromosome) mutations, the risk of ovarian cancer begins to rise in their late 30s and is estimated to result in a lifetime risk of 25% to 40% (Hennessy et al. 2009). Women who carry BRCA2 (13q12 chromosome) gene mutations also are at substantial risk of developing ovarian cancer (15%–25%). BRCA1 and BRCA2 are tumor suppressor genes inherited in an autosomal dominant mode (Schmeler et al. 2006).
Spread of ovarian cancer beyond the ovary occurs in three ways. First, the tumor can penetrate the ovarian capsule and directly invade contiguous organs. Second, the tumor cells may spread via the lymphatics involving the pelvic and para-aortic lymph nodes. Lymph node metastases occur in as many as 20% of early stage cancers and the majority of advanced-stage cancers. Third, ovarian cancer cells will escape from areas where the tumor has penetrated the capsule of the ovary and escape into the peritoneal cavity. This latter form of spread is the most devastating because it results in the dissemination of tumor cells throughout the peritoneal cavity (Bharwani et al. 2011). Stage I tumor is limited to the ovaries, stage II tumor involves one or both ovaries with pelvic extension, stage III tumor involves one or both ovaries with confirmed peritoneal metastasis outside the pelvis and/or regional lymph node metastasis, and stage IV tumor is accompanied by distant metastasis (Bhoola and Hoskins 2006).
Approximately 90% of primary malignant ovarian tumors are epithelial in origin (carcinomas). The remainder are germ cell or stromal tumors. Epithelial ovarian carcinomas are thought to arise from the ovarian surface epithelium or from surface epithelial inclusion cysts. Ovarian carcinomas are heterogeneous and are primarily classified by cell type into serous, mucinous, endometrioid, clear cell, and Brenner (transitional) tumors corresponding to different types of epithelia in the organs of the female reproductive tract. Serous tumors are the most common type of ovarian epithelial tumor, accounting for about 53% of cases (Seidman and Kurman 2003). The tumors in each of the categories are further subdivided into three groups—benign, malignant, and intermediate (borderline tumor)—to reflect their behavior.
The pathogenesis of ovarian carcinoma is unknown because of the lack of a tumor progression model. Based on clinical, morphologic, and molecular genetic characteristics, a model of ovarian carcinogenesis has been proposed. In this model, there are two main pathways of tumorigenesis. In this model, type I and type II refer to pathways of tumorigenesis and are not specific histopathological terms (Shih and Kurman 2004). Type I tumors include low-grade serous carcinoma, mucinous carcinoma, endometrioid carcinoma, malignant Brenner tumors, and clear cell carcinoma. Type II tumors are composed of what are currently classified as moderately and poorly differentiated serous carcinoma (high-grade serous carcinoma), malignant mixed mesodermal tumors (carcinosarcomas), and undifferentiated carcinoma. The tumorigenic pathway for type I tumors resembles the adenoma-carcinoma sequence in colorectal cancer. They tend to present as stage I, low-grade neoplasms that develop in a stepwise manner from well-recognized precursors—namely, borderline tumors that in turn develop from cystadenomas and adenofibromas. They behave in an indolent fashion (Bell DA 2005).
Type I tumors evolve slowly and are associated with distinct molecular changes that are rarely found in type II tumors. These low-grade tumors are relatively genetically stable and are characterized by mutations in a number of genes. The best characterized molecular alterations are sequence mutations in KRAS, BRAF, and ERBB2 oncogenes. Previous studies have demonstrated that KRAS mutations occur in 35% of low-grade serous carcinomas and 33% of borderline tumors but not in high-grade serous carcinomas. Similarly, BRAF mutations occur in 30% of low-grade serous carcinomas and 28% of borderline tumors but not in high-grade serous carcinomas (Singer et al. 2003). Mutations in ERBB2 occur in less than 5% of these tumors. Mutations in KRAS, BRAF, and ERBB2 are mutually exclusive. Therefore, mutations in these genes are detected in about two-thirds of type I tumors. The most common molecular genetic alteration in mucinous tumors is point mutations of KRAS (Gemignani et al. 2003). In endometrioid carcinomas, mutation of β-catenin has been reported in approximately one-third of cases and mutation of PTEN in 20%. PTEN mutations rising to 46% have been seen in those tumors with 10q23 loss of heterozygosity (Kim et al. 2008).
In contrast, type II tumors evolve rapidly. They nearly always present as high-stage, high-grade tumors that metastasize early in their course and are extremely aggressive (Kurman et al. 2008). Type II carcinomas are rarely associated with morphologically recognizable precursor lesions, and it has been proposed that they develop de novo from the surface epithelium or inclusion cysts of the ovary (Bell KA and Kurman 2000). The only mutation that has been consistently detected in type II tumors are mutations in TP53, which are very rare in type I tumors. In addition, type II tumors are characterized by considerable genetic instability, which is not observed in type I tumors.
Heparan Sulfate: A Very Complex Carbohydrate
All mammalian tissues contain heparan sulfate, and virtually all cells synthesize this glycosaminoglycan. The only notable exception is erythrocytes. Therefore, given the ubiquitous distribution of this carbohydrate and its ability to bind and concentrate many tumor-related growth factors, cytokines, and morphogens (Fuster and Esko 2005), as well as regulate cell adhesion and migration through association with adhesion receptors, interest in heparan sulfate has intensified over the past decade.
Heparan sulfate is synthesized in the Golgi apparatus and always attached to a core protein; it is apparently never secreted as a free glycosaminoglycan. A variety of core proteins can bear heparan sulfate chains, covalently attached through specific serine residues. Many occur on the cell surface and are represented mainly by two families, the transmembrane syndecans and glycosylphosphatidylinositol (GPI)–anchored glypicans. Both are ancient families, represented in invertebrates, including Caenorhabditis elegans and Drosophila (Couchman 2003, 2010; Esko and Selleck 2002; Kirkpatrick and Selleck 2007; Lin X 2004; Mythreye and Blobe 2009). In addition, there are basement membrane and matrix heparan sulfate proteoglycans (HSPGs), such as perlecan, type XVIII collagen, and agrin (Iozzo et al. 2009). Heparan sulfate is a highly complex carbohydrate, perhaps the most complex known. The stem structure of xylose–galactose–galactose–uronic acid (from reducing terminus) is then followed by repeating disaccharides of glucosamine and uronic acid (Figure 1). The complexity lies in subsequent modifications of the chains. These are sequential and commence with N-deacetylation and N-sulfation of the glucosamine residues. Some of the adjacent uronic acid residues are then epimerized from glucuronic to iduronic acid, of which many are then 2-O-sulfated. Then follow 6-O-sulfation and, to a lesser extent, 3-O-sulfation of the hexosamine residues to yield mature chains. However, for many proteoglycans, none of these modification proceeds to completion. The result is a chain that has regions of low sulfation and modification, alternating with regions of higher sulfation, often with intermediate sulfation at the boundaries of the subdomains (Murphy et al. 2004). It is the higher sulfated regions that interact with peptide ligands, such as growth factors and extracellular matrix molecules. It seems clear that the pattern of heparan sulfate modification is not random but controlled by the cell, although how the transferases are organized in the Golgi apparatus to achieve this complexity is unknown. This is all the more impressive given that there are 12 different possible sulfated disaccharides (Karamanos et al. 1996). Moreover, in mammalian cells, there are multiple transferase genes encoding proteins with ostensibly the same function. For example, there are four N-deacetylase/N-sulfotransferase genes, but they exhibit tissue-specific expression and show little redundancy (Aikawa et al. 2001). Understanding the regulation of heparan sulfate synthesis remains a large challenge. There have been a few in-depth recent reviews of this area recently (Kreuger et al. 2006; Lindahl and Li 2009; Skidmore et al. 2008).
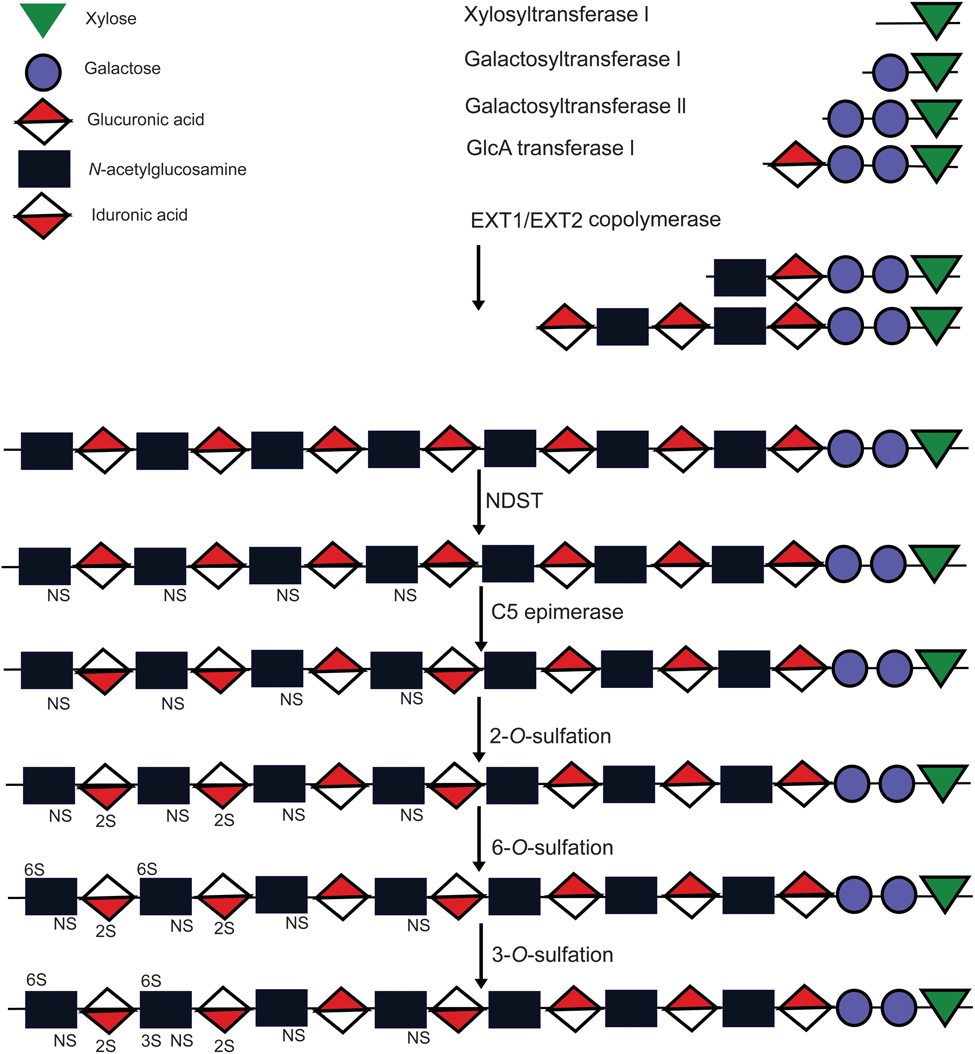
Heparan sulfate (HS) biosynthesis. HS synthesis involves sequential addition of N-acetylglucosamine and glucuronic acid (GlcA) residues to a tetrasaccharide linker region, which is covalently linked to the serine residue within the core protein GAG attachment site. Each sugar residue is depicted by a geometric symbol defined in the legend at the top left corner of the figure. The chain then undergoes stepwise modifications beginning with N-deacetylation and N-sulfation of some glucosamine residues. Adjacent to this first modification, some GlcA residues are epimerized to iduronic acid by a 5′ epimerase. Iduronate may then be 2-O-sulfated, with further sulfation on 6-O and 3-O positions on glucosamine residues. None of these modifications goes to completion, leading to a domain structure of HS chains with regions that are highly sulfated, flanked by regions of intermediate sulfation and low sulfation. Much more extensive chain modification is seen in the synthesis of the skeletal polysaccharide, heparin.
Further complexity arises on the cell surface. Both sulfatases and mammalian heparanase can be widespread. The former are specific for 6-O-sulfate residues of glucosamine, the latter cleaving the chains. Both affect cell behavior. Several studies have shown the significance of these enzymes on cellular responses, and both have been implicated in tumor progression (Arvatz et al. 2011). Because some growth factor interactions and productive signaling can depend on specific sulfation motifs, it is not surprising that sulfatases can specifically alter some of these (Morimoto-Tomita et al. 2005). Heparanase can be upregulated in a number of tumor cell types, again raising the prospect of altered cell behavior and a significant contribution to pathogenesis. From their detailed studies of heparan sulfate interactions with growth factors, Backen et al. (2007) estimated that the activity of editing enzymes is of major significance in ovarian cancer.
Mammalian Syndecans: Multitasking Receptors
Syndecans are in some ways typical type I membrane receptors, with the added complication of attached glycosaminoglycan chains (Couchman 2010; Morgan et al. 2007). These are commonly heparan sulfate, and it is believed currently that syndecan core proteins are always substituted with at least one form of glycosaminoglycan (Figure 2). Heparan sulfate chains can interact with a large array of ligands from many different classes of extracellular protein, including growth factors, morphogens, cytokines and chemokines, extracellular matrix molecules, clotting factors, and enzymes such as thrombin, metalloproteinases, and lipases. Techniques are not yet available to determine under what circumstances ligands bind in vivo, but despite what appears to be a complete lack of ligand specificity, knockouts of some transferases involved in heparan sulfate synthesis have specific and often lethal phenotypes (Bishop et al. 2007). Heparan sulfate is required for multicellular animal life.
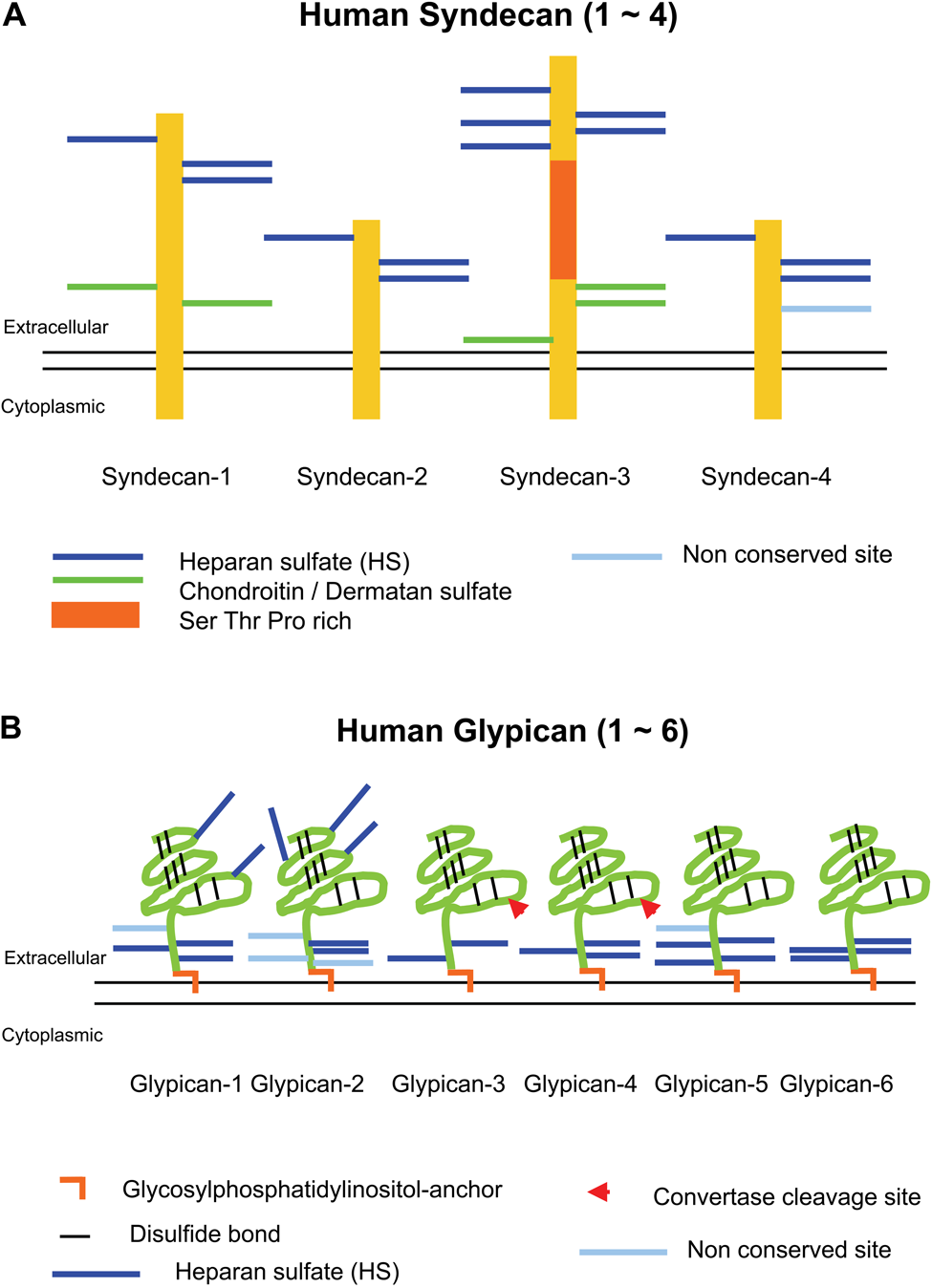
Diagram of syndecans and glypicans. Schematic representations of human syndecan 1–4 monomers (A) and human glypican 1–6 (B) are shown.
The external core proteins, on which glycosaminoglycans are substituted, do not exhibit strong sequence homology between the four syndecans or even between species for the same syndecan. This led to an early speculation that the protein was nothing more than a carrier of carbohydrate chains, but this has had to be revised. For syndecan-1, -2, and -4 at least, the external core protein can trigger integrin-mediated cell adhesion events, which may be direct or, in the case of syndecan-4, probably indirect (Couchman 2010). The mechanism of integrin involvement still remains to be elucidated, but conceivably this may happen in cis on the cell surface or in trans from an adjacent cell or, since syndecans can be shed, mediated by a soluble shed form. Syndecan shedding has been the subject of a recent review (Manon-Jensen et al. 2010).
Cell adhesion comes to the fore again when considering cytoplasmic domains. Each of the four syndecans has been proposed to connect to the actin cytoskeleton (Couchman 2003, 2010), through ezrin in syndecan-2 and α-actinin in the case of syndecan-4, to Src and its substrate, cortactin, in the case of syndecan-3. The cytoplasmic domains can be divided into three regions, two highly conserved in sequence across species and across the syndecans. The C1 region is membrane proximal, whereas C2 is distal and represents the carboxyl terminus. In between is a variable (V) region that is different across the syndecan members yet quite conserved across species within each syndecan. Likely, there are shared properties of the conserved (C) regions, whereas each V region has syndecan-specific functions. A small number of class II PDZ proteins can interact with C2 regions of syndecans (Multhaupt et al. 2009), the most studied being syntenin and others being synectin (GIPC), synbindin, and CASK. Syntenin appears capable of binding all syndecans and may be involved with their intracellular trafficking (Zimmermann et al. 2005).
Signaling through Syndecans
The V regions of syndecans have been difficult to study, and this is perhaps because all syndecans form stable dimers, mediated in part by a GXXXG motif found within all syndecan transmembrane domains (Dews and Mackenzie 2007). Furthermore, the cytoplasmic domains of syndecan-4 have been shown by NMR spectroscopy to form an unusual twisted clamp dimer, which is stabilized by interactions with the phospholipid PtdIns 4,5 P2 (Lee et al. 1998). In turn, this forms a complex with protein kinase Cα, which is then persistently activated (Keum et al. 2004) and signals downstream to G proteins such as Rac and RhoA and on to rho kinases and actomyosin (Couchman 2010; Morgan et al. 2007). Syndecan-4 can therefore influence actin cytoskeletal architecture, and consistent with this, syndecan-4 can be incorporated into focal adhesions along with essential integrins (Baciu and Goetinck 1995; Couchman 2010; Morgan et al. 2007; Woods and Couchman 1994). Focal adhesions are important signaling organelles, involved with not only tight cell-extracellular matrix adhesion but also cell survival (Dubash et al. 2009). Experiments with Chinese hamster ovary (CHO) cells have shown that overexpression of syndecan-4 can lead to increased focal adhesion assembly and reduced migration as a result (Longley et al. 1999). It has long been known that focal adhesions, being points of stable cell-matrix interaction, oppose cell migration. However, fibroblasts from syndecan-4 knockout mice are also migration-defective in vitro and possibly in vivo because granulation tissue angiogenesis in skin wound healing is reduced (Echtermeyer et al. 2001). It therefore seems as if efficient cell migration requires enough syndecan-4 but not too much. Elegant studies have also shown that syndecan-4 is also required for directional persistence of migration, a Rac-dependent process (Bass et al. 2007). The underlying signaling control of these processes is not yet fully understood. However, by comparison, very little is understood about the other syndecan cytoplasmic domains, perhaps because oligomerization and structural constraints have to be satisfied before signaling complexes can be assembled. This is a challenge that needs to be met. Certainly, the connection between cell migration and possibly invasion may be a potent component of tumor cell progression.
The Glypicans
The glypican family comprises six members (glypican 1–6) in human and mouse and two members (Dally and Dally-like protein [Dlp]) in Drosophila. The glypicans can be further divided into two groups based on sequence homology: (1) glypicans 1, 2, 4, and 6 and Dlp and (2) glypicans 3 and 5 and Dally (Filmus et al. 2008). This family of cell surface HSPG is anchored through glycosylphosphatidylinositol in the outer layer of cell membranes. It has been shown that some members of glypicans, including glypican-3, can be processed by furin-like convertase, which favors binding of Wnt (De Cat et al. 2003; Filmus et al. 2008). Glypican can be detected in conditioned medium of cultured cells, but unlike syndecans, there are no reports of glypican shedding by proteases. Instead of shedding by proteases, glypicans can be released from cell membranes by the extracellular lipase Notum at the GPI anchor (Kreuger et al. 2004). Heparan sulfate attachment sites on glypicans are predominantly located close to the cell membrane. There are no similarities in structure between syndecans and glypicans, at least based on sequence comparisons. Glypicans usually have eight cysteine residues, indicative of a globular structure with four disulfide bridges.
Genetic interaction experiments in Drosophila show that glypicans regulate bone morphogenetic protein (BMP), fibroblast growth factors (FGF), hedgehogs (Hh), and wingless (Wg), all of which have heparin-binding properties (Filmus et al. 2008). Glypicans seem to regulate morphogen gradients in the extracellular matrix and the interaction of cytokines with other receptors by facilitating and/or stabilizing them. The contribution of glypican in FGF signaling is mainly mediated by heparan sulfate chains (Song et al. 1997). However, molecular properties of glypicans other than heparan sulfate seem to be important for Wnt (Song et al. 2005), Hh, and BMP because glypican-3 core protein can also interact with Hh (Capurro et al. 2008).
In one case, glypican-3, there is a clear connection to a human genetic disease, Simpson-Golabi-Behmel syndrome (Pilia et al. 1996). This is characterized by overgrowth, strongly suggesting that growth factor metabolism is altered, most likely that of Hh (Capurro et al. 2008). In many cases, mutations lead to a secretion failure; the misfolded proteins accumulate intracellularly. Patients with this disease are prone to tumors, notably Wilms tumors of the kidney. The latest data suggest that the molecular basis for the disease lies in disrupted hedgehog signaling. Therefore, in Simpson-Golabi-Behmel syndrome, inappropriate Hh metabolism may lead to the overgrowth and tumor susceptibility. Certainly, the disease shows that there is no functional redundancy with respect to glypican-3 at least, because no other glypican can substitute.
Most evidence suggests that glypicans are important in growth factor and morphogen responses, whereas roles in cell adhesion seem to be the prerogative of syndecans. In Drosophila, there are two glypican genes and one syndecan (Kirkpatrick and Selleck 2007; Couchman 2010). Deletions and mutations in the glypicans Dally and Dally-like influence a number of growth factors (e.g. FGF, Wg, and Hh), whereas syndecan’s role in invertebrates has been shown to relate particularly to neural path finding and the Slit-Robo system (Lin X 2004; Steigmann et al. 2004).
Glypicans in Breast and Ovarian Cancers
Because many growth factors are implicated in tumor progression and tumor angiogenesis, for example, it is not surprising that changes in glypican expression have been noted in several human tumors. Among the glypican family, GPC3/OCI-5/MXR7 has been well studied regarding cancer biology. The GPC3 gene is located at Xq26, a region frequently deleted in advanced ovarian cancers (Choi C et al. 1998). Silencing of GPC3 gene is reported in ovarian cancer (Lin H et al. 1999) and human breast cancer (Xiang et al. 2001). The mechanism of GPC3 silencing was due, in part, to hypermethylation of the GPC3 promoter, especially in hormone receptor–negative tumors (Yan et al. 2001). Ectopic expression of glypican-3 inhibited growth in 8 of 10 breast cancer cell lines (Xiang et al. 2001). Other research reported that this proteoglycan induced apoptosis in breast cancer MCF7 cells but not in colorectal tumor cells, although heparan sulfate was not required for this activity (Gonzalez et al. 1998). With ectopic expression of glypican-3 in the LM3 mammary tumor cell line, its contribution to increased apoptosis sensitivity induced by serum depletion was noted (Peters et al. 2003). Collectively, these data suggest that glypican-3 can act as a negative regulator of breast cancer growth. In the case of ovarian cancer, expression of this HSPG was restored by treatment of cells with a demethylating agent, whereas ectopic glypican-3 expression inhibited ovarian cancer cell growth (Lin H et al. 1999).
In contrast, glypican-1 is strongly expressed in human breast cancers but low in normal breast tissues (Matsuda et al. 2001). In the same study, it was found that the expression of glypican-3 and -4 was only slightly increased in cancers by comparison with normal tissues, and glypican-2 and -5 were below the level of detection in both normal and cancer samples. Treatment of breast tumor cells MDA-MB-231 and MDA-MB-468 with phosphoinositide-specific phospholipase C to release glypicans abrogated the mitogenic response to two heparin-binding growth factors, HB-EGF and FGF2 but not IGF-1. Reduction of glypican-1 protein level markedly decreased the mitogenic response to these growth factors as well as that to heregulin α, heregulin β, and hepatocyte growth factor (HGF). However, loss of mitogenic responsiveness to these growth factors in these clones was not due to altered syndecan-1 mRNA expression. A study by Lander’s group (Ding et al. 2005) may resolve the molecular mechanism in the case of FGF2. Using pancreatic cancer cells, it was shown that treatment of cancer cells with FGF2 induced shedding of syndecan-1 and converted the FGF2 response to glypican dependent. This shedding was observed in the breast carcinoma MDA-MB-468 line as well. In pancreatic cancer cells, syndecan-1 shedding was MMP-7 dependent, which is a heparin-binding molecule itself and anchored to cells by HSPG (Ding et al. 2005). It was also reported that ectopic expression of glypican-3 in mammary tumor cells also prevented the increase of MMP-2 induced by insulin-like growth factor II (IGF-II) (Peters et al. 2003).
The same trend with both syndecan-1 and glypican-1 has been recorded for ovarian cancer, and their upregulation is a poor prognostic factor. This also applies to stromal localization of the two proteoglycans (Davies et al. 2004). The relevance of other glypican members in ovarian carcinoma is not well understood. One report indicates that glypican-3 is absent or undetectable in the vast majority of clear cell, serous, endometrioid, and mucinous tumors (Esheba et al. 2008). A second report, however, suggested that around 18% of ovarian carcinomas were glypican-3 positive, these being of the clear cell type (Stadlmann et al. 2007). However, even where expressed, there was no association with tumor stage. Positive staining for this glypican was noted in a proportion of recurrent cases but not associated with chemoresponse.
Clear cell carcinoma occurs more frequently in Japan than in Western countries, and two early studies with small numbers of cases yielded conflicting results regarding glypican-3 expression in clear cell adenocarcinoma of the ovary (Stadlmann et al. 2007; Esheba et al. 2008). However, more recent studies focusing on clear cell carcinoma have been performed in Japan. GPC3 was expressed in about 40% of clear cell carcinomas (Maeda et al. 2009; Umezu et al. 2010) but rare in other types such as mucinous, endometrioid, or serous (Maeda et al. 2009). One study showed that glypican-3 was significantly associated with poor overall survival in stage III/IV clear cell adenocarcinoma cases (Maeda et al. 2009), whereas another study showed association with poor progression-free survival in stage I clear cell carcinoma patients (Umezu et al. 2010). Both studies showed no correlations between its expression and clinicopathological factors (Maeda et al. 2009; Umezu et al. 2010). Stromal expression of glypican-1 was associated with a poor prognosis in ovarian cancers (Ricciardelli and Rodgers 2006; Davies et al. 2004).
Syndecans in Breast and Ovarian Carcinoma
Most data relating to syndecans in these tumors concern syndecan-1. This proteoglycan has a predominantly epithelial distribution, so it has been studied in many carcinomas. Upregulation at the protein and mRNA levels has been recorded in breast oncogenesis and correlates with higher histological tumor grade, as well as increased mitotic index and tumor size, and therefore is a poor prognostic factor (Lendorf et al. 2011). This last feature is common with ovarian carcinoma, but it is not universally true for all tumor types. Decreased expression and possible inhibitory functions have been suggested in lung, colorectal, and cervical cancers (Antonnen et al. 2001; Fujiya et al. 2001; Lundin et al. 2005; Shinyo et al. 2005). In the Wnt-1 expression model of mammary tumorigenesis in the mouse, syndecan-1 was shown to be essential because knockout mice were resistant (Alexander et al. 2000). This suggests a role for syndecan-1, either directly or indirectly, in Wnt signaling, but it is also interesting that in this model, there is no redundancy. Presumably, syndecan-4 and some glypicans were present in the mammary epithelia but were unable to substitute for syndecan-1.
Syndecan-4 is also present in mammary epithelium but at lower levels than syndecan-1. It has not been intensively studied in either breast or ovarian cancer, and there are some differences in conclusions. One report shows that syndecan-4 and -1 are similar in breast cancer tissue, with increased expression being a poor prognostic indicator (Baba et al. 2006). However, a second report suggests downregulation of syndecan-4 in malignant breast tissue (Mundhenke et al. 2002). Our own study suggests that there is not a correlation between syndecan-4 expression and tumor type or grade. Rather, there was a correlation with expression of estrogen and progesterone receptor status (Lendorf et al. 2011). Given that treatment options for patients with receptor-positive status are wider, this could indicate a better prognosis. We also showed that stromal syndecan-4 was a rare feature across all breast carcinomas, unlike syndecan-1, which had a stromal expression that was strongly associated with a higher tumor grade (Lendorf et al. 2011). Examples of syndecans-1 and -4 in ovarian and breast carcinoma are shown in Figures 3 and 4.
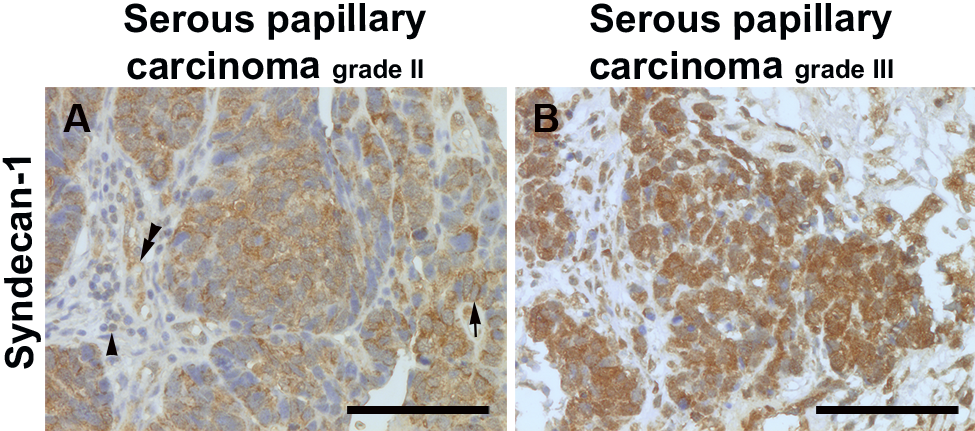
Ovarian carcinoma immunostained for syndecan-1. Serous papillary carcinomas of the ovary, grade II (A) and grade III (B), were stained with mouse monoclonal antibodies against human syndecan-1. (A) The grade II tumor cells strongly express syndecan-1, as shown by the positive cytoplasm. Focal membranous staining is also visible (arrow). Note syndecan-1-positive endothelial cells in the blood vessels (double arrowheads) and positive infiltrating immunological cells in the tumor stroma (arrowhead). (B) Tumor cells of grade III ovarian serous carcinoma show overall strong cytoplasmic and focally nuclear staining for syndecan-1. Scale bar: 100 µm.
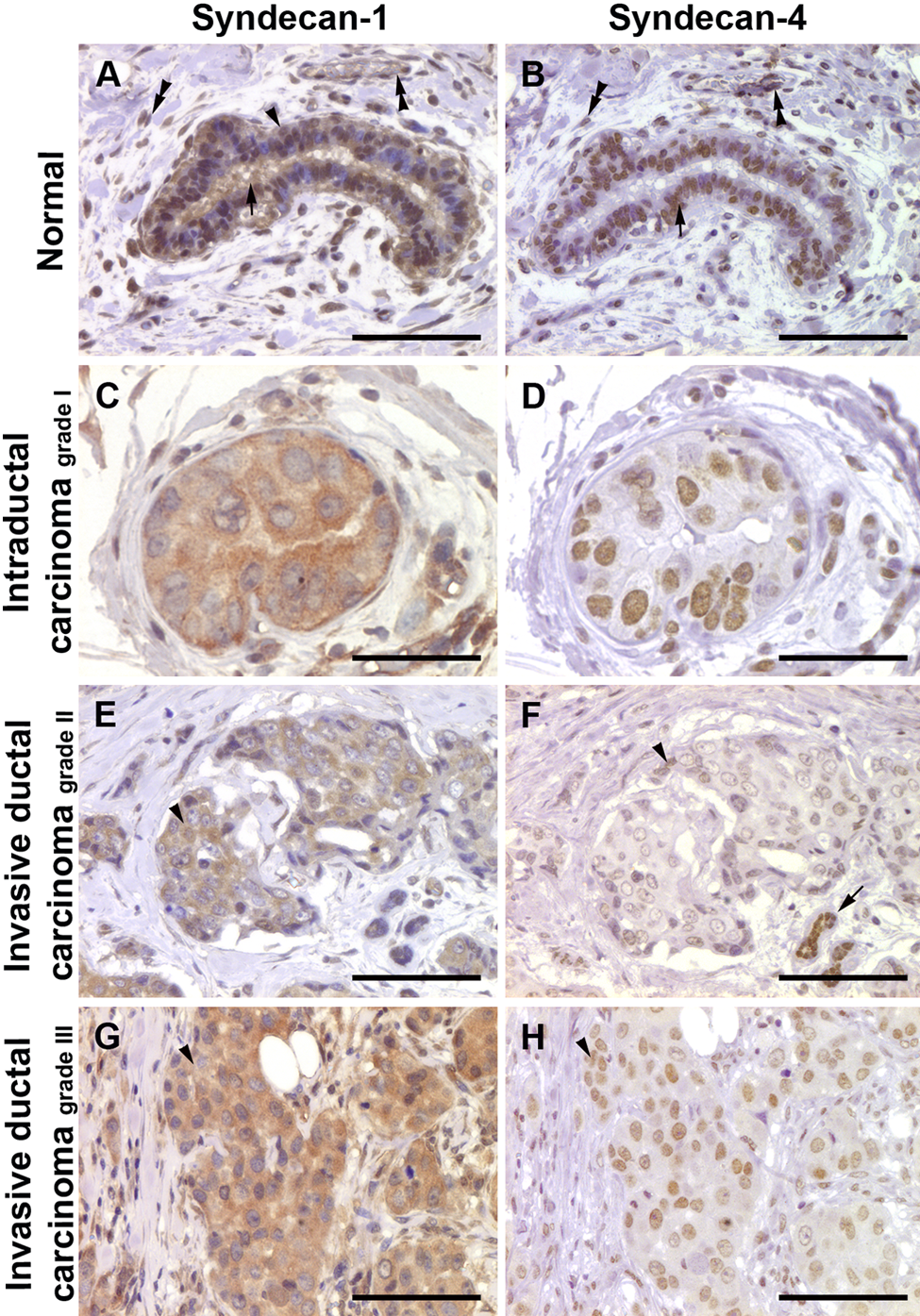
Immunohistochemical staining of human breast tissues for syndecan-1 and syndecan-4. Tissue microarray containing cores of normal human breast (A and B; presented case is AR++, ER+, PR++, and Her2++), intraductal carcinoma (C and D; presented case is AR–, ER+++, PR–, and Her2+++), and invasive ductal carcinoma grade II (E and F; presented case is AR–, ER+, PR–, and Her2+++) and grade III (G and H; presented case is AR–, ER–, PR–, and Her2–) were stained with mouse monoclonal antibodies against human syndecan-1 and syndecan-4. (A) Normal human breast ducts show positive syndecan-1 staining in myoepithelial (arrowhead) and secretory cells (arrow). The positive staining is cytoplasmic as well as nuclear. Endothelial cells of the blood vessels as well as the stromal fibroblasts are also positive (double arrowheads). (B) Syndecan-4 staining shows a predominant nuclear staining pattern (arrow); in addition, the stromal fibroblasts and endothelial cells are also positive (double arrowheads). (C, D) Intraductal carcinoma of the breast has strong cytoplasmic expression of syndecan-1 (C), whereas the syndecan-4 staining is nuclear (D). (E) Invasive ductal carcinoma grade II tumor cells show cytoplasmic expression for syndecan-1 (arrowhead). (F) The tumor cells only show very weak nuclear staining for syndecan-4 (arrowhead); the normal ducts are strongly positive (arrow). (G) Invasive ductal carcinoma grade III shows positive syndecan-1 staining in the cytoplasm (arrowhead). (H) Syndecan-4 shows a nuclear staining pattern (arrowhead). Scale bar: 100 µm (A, B, E–H); 50 µm (C, D).
Syndecan-3 has not been reported widely with respect to either breast or ovarian carcinoma, but its aberrant upregulation in ovarian carcinoma-associated vasculature has been noted (Davies et al. 2004). Syndecan-2, on the other hand, has been studied more extensively and its expression reported in ovarian carcinoma and several other tumor types such as lung cancer, osteosarcoma, mesothelioma, colorectal cancer, and brain tumors (Theocharis et al. 2010). It has also been reported that its expression in breast carcinoma is regulated by estradiol, through the action of estrogen receptor α (Kousidou et al. 2008). In particular, this syndecan has been suggested to be a promoter of tumor angiogenesis, and it may be strongly expressed in vascular tissues (Fears et al. 2006). Several tissue culture studies also suggest that colon carcinoma cell expression of syndecan-2 leads to Rac-related increased invasive behavior (Park et al. 2002; Choi Y et al. 2010), perhaps through an ability to interact with many tumor progression–promoting growth factors. However, it is once again dangerous to generalize. Studies with osteosarcoma cells suggest that syndecan-2 may be inhibitory and promote cell death through apoptosis (Modrowski et al. 2005).
Mutations in Syndecan and Glypican Core Proteins or in Enzymes That Contribute to Heparan Sulfate Synthesis and Editing
A number of mutations related to proteoglycans have been recorded in breast and ovarian cancer (Table 1). Most have been identified in ovarian serous carcinoma, and the majority of these mutations lead to changes in amino acid sequence. These mutations involve not only the core proteins of syndecans and glypicans but also some enzymes that are involved in heparan sulfate synthesis or modification of the mature polysaccharide. Four mutations have been described in syndecan-1 and two in syndecan-2 from ovarian carcinoma patients. A single mutation in syndecan-3 core protein is presumably silent because no change in amino acid sequence would follow. The functional consequences of the syndecan-1 and -2 mutations are unknown currently but may influence expression. One mutation in syndecan-1 that predicts a glycine 36→alanine replacement is immediately N-terminal of the first serine-glycine dipeptide at which heparan sulfate chain attachment occurs. Substitution of heparan sulfate chains always occurs at these serine-glycine motifs, which are usually surrounded by acidic residues (Couchman 2003; Skidmore et al. 2008). The amino acid change is conservative, however, so it is unlikely to influence glycanation. Similarly, the valine 25→leucine mutation in syndecan-1 may not have much effect, being apparently a conservative change within the signal sequence. The consequence of amino acid changes at residues 154 and 155, located around midway in the ectodomain, is unclear because of a dearth of structural studies on syndecan ectodomains, but residue 155 is not strongly conserved across mammals.
Mutations in Genes Encoding Heparan Sulfate Proteoglycan Core Proteins and Heparan Sulfate Synthesis and Modification
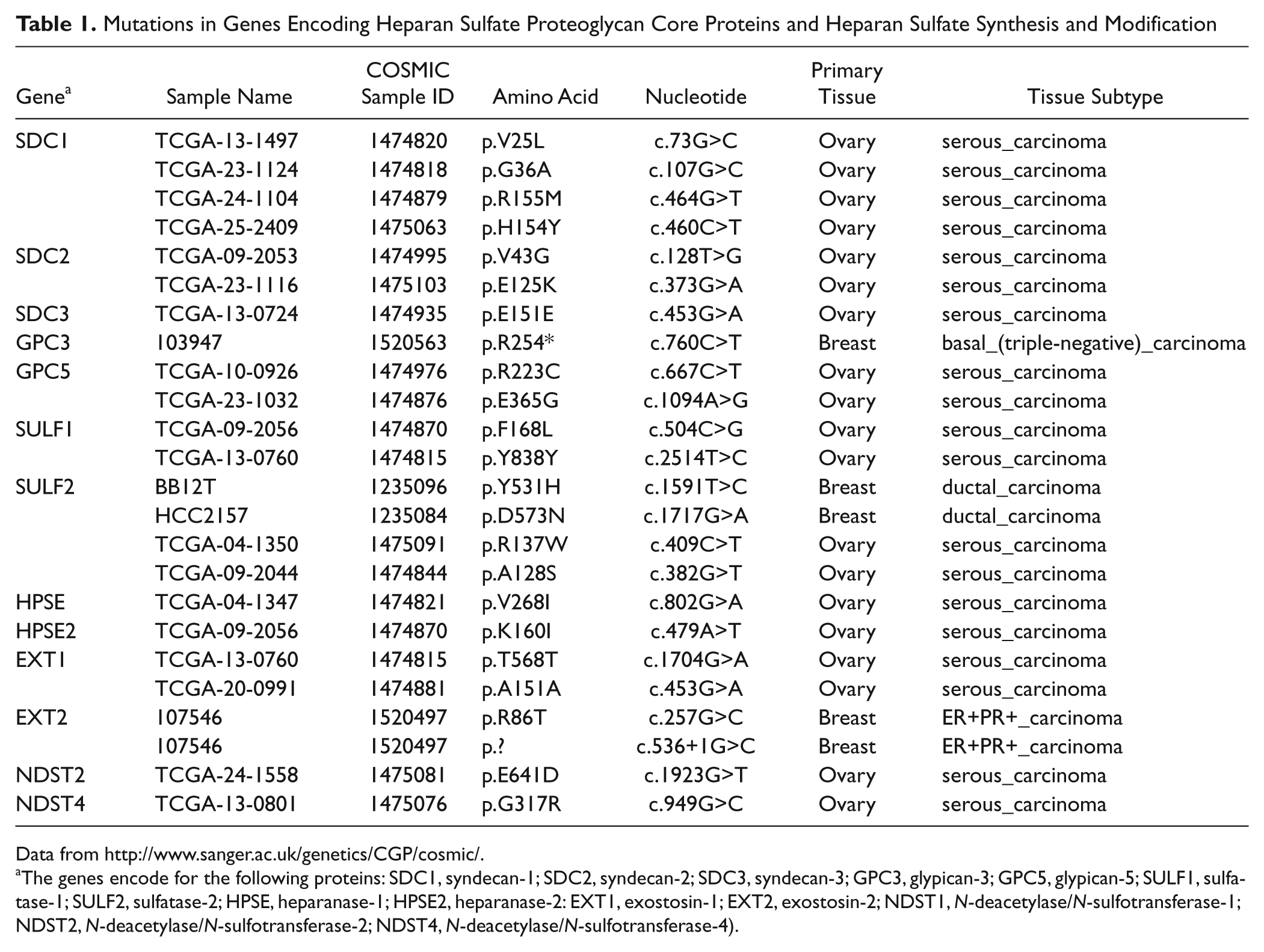
Data from http://www.sanger.ac.uk/genetics/CGP/cosmic/.
The genes encode for the following proteins: SDC1, syndecan-1; SDC2, syndecan-2; SDC3, syndecan-3; GPC3, glypican-3; GPC5, glypican-5; SULF1, sulfatase-1; SULF2, sulfatase-2; HPSE, heparanase-1; HPSE2, heparanase-2: EXT1, exostosin-1; EXT2, exostosin-2; NDST1, N-deacetylase/N-sulfotransferase-1; NDST2, N-deacetylase/N-sulfotransferase-2; NDST4, N-deacetylase/N-sulfotransferase-4).
The glutamate→lysine mutation in syndecan-2 reverses the charge at residue 125, which may have effects on folding or interactions. The replacement of valine with glycine at residue 43 of syndecan-2 is interesting because this residue immediately follows the first serine-glycine heparan sulfate substitution site. Whether it affects glycanation can be determined experimentally, but again this residue is not conserved, being replaced by leucine and alanine in rat and chicken, respectively. Most likely, there is no effect on heparan sulfate attachment to the core protein.
Of potential interest, two cases of glypican-5 mutation in ovarian serous carcinoma are now recorded. These mutations are in the central region of the core protein, and in one case, the mutation creates an unpaired cysteine residue, which may have a significant impact on the folding of the molecule and, perhaps, its interactions. It could be relevant to know whether these mutations affect expression on the cell surface, half-life, glycanation, or other functional attributes. There is also a single case of a mutation in glypican-3 in ductal carcinoma of the breast. This predicts a premature stop codon, and because this proteoglycan is known in other diseases to influence proliferation (Capurro et al. 2008), this mutation may be important to study further.
The remaining mutations relevant to proteoglycans relate to heparan sulfate synthesis or its subsequent modification. EXT1 and 2 form a heterodimeric complex that is the major polymerase responsible for heparosan chain assembly. Two mutations from ovarian carcinoma in EXT1 are apparently silent, but that is not the case with the two EXT2 mutations identified in breast carcinoma patients (Table 1). After chain assembly, many of the N-acetylglucosamine residues are modified by a combination of N-deacetylation and N-sulfation (Fig. 1). Of the four human enzymes that carry out this dual function, single mutations in ovarian carcinoma are known in NDST2 and NDST4. The former leads to a conservative change in acidic residue, whereas the latter replaces a glycine with arginine. Whether this affects activity is unknown, but NDST4 is quite tissue restricted in distribution, and in many tissues, NDST1 is the major enzyme in terms of expression levels.
The sulfatases can selectively remove 6-O-sulfate groups from glucosamine residues, and Sulf2, in particular, is under scrutiny with respect to the progression of several tumor types (Rosen SD and Lemjabbar-Alaoui 2010). This sulfatase is known to be upregulated in breast cancer (Morimoto-Tomita et al. 2005). The effect of enzyme activity is to modify cellular responses to certain growth factors. Two mutations in Sulf1 and four in Sulf2 are known from breast and ovarian carcinomas, and in every case but one, this leads to changes in amino acids. Finally, heparanases are also under investigation in tumor biology because they cleave heparan sulfate chains to release bioactive oligosaccharides that may influence cell behavior (Vlodavsky et al. 2011). Two mutations from ovarian carcinoma are recorded, but as with the Sulf mutations, what influence the amino acid substitutions have on enzymatic activity is unknown but potentially relevant to pursue.
Conclusions and Perspectives
In both breast and ovarian carcinoma, there are changes in the expression and distribution of syndecans and glypicans. In some cases, these changes correlate significantly with poorer outcome. For example, in cases where syndecan-1 is present in the stroma of mammary tumors, there is a clear linkage to disease progression and tumor grade. Where this stromal syndecan-1 originates is not entirely clear, although likely it is of several sources, including the activated cancer-associated fibroblasts. Virtually nothing is known regarding possible alterations in heparan sulfate fine structure that may accompany these changes in proteoglycan expression. This may be relevant, given heparan sulfate’s ability to interact with many potent growth-promoting polypeptides.
Is the elevated presence of a proteoglycan, such as glypican-1 or syndecan-1, in tumors of significance as a contributor to tumor progression or merely an effect of elevated growth and increased invasive behavior? The answer is not clear, but the possible presence of mutations in syndecans and glypican-5 in ovarian cancer is intriguing. Moreover, syndecan-4 is known to be upregulated in cells bearing KRAS mutations, as is often seen in ovarian tumors (Bild et al. 2006). It is tempting to believe that elevated levels of cell surface heparan sulfate are contributors to pathogenesis because their ability to interact with mitogenic peptides and act in co-receptor roles with high-affinity tyrosine kinase receptors puts this carbohydrate at a crucial place in cell communication. If we look across to myeloma, the evidence for a critical role of syndecan-1 seems certain (Sanderson and Yang 2008). Circulating levels of shed syndecan-1 correlate with tumor progression, and experimental downregulation of the core protein led to much reduced proliferation of the tumor cells. It provides hope that a syndecan may form the basis of a therapeutic target for this devastating disease. In turn, these data suggest that cell surface proteoglycans in other tumors, including those highlighted here, may be important and relevant for further work. One key factor in their favor is that they are on the cell surface and are accessible targets. Only further work, particularly with animal models, will answer these key questions.
Footnotes
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
The authors disclosed receipt of the following financial support for the research and/or authorship of this article: Support from The Danish National Research Foundation (HABM, JRC), Fabrikant Vilhelm Pedersen og Hustrus Legat through Novo Nordisk Fonden, Lundbeck Fonden, The Department of Biomedical Sciences at The University of Copenhagen (JRC), and Danish Cancer Research Foundation (AY) is gratefully acknowledged. MEL was supported by a Rota System PhD Fellowship from the Copenhagen Graduate School of Health Sciences.