
Abstract
Tartrate-resistant acid phosphatase (TRACP) is a cytochemical marker for hairy cell leukemia, macrophages, dendritic cells, and osteoclasts. Our purpose was to develop multicolor cytofluorometric methods to evaluate intracellular TRACP enzymic activity using a fluorogenic cytochemical reaction in combination with immunochemical stains for distinct surface membrane antigens. Monocyte-derived dendritic cells (DCs) were the model TRACP-expressing cells studied. Intracellular TRACP activity was disclosed using naphthol-ASBI phosphate as substrate with fast red-violet LB salt as coupler for the reaction product. Before the TRACP enzymic reaction, surface antigens, CD86 and CD11c of DCs, were bound with specific fluorescent antibodies to test compatibility of surface labeling and intracellular staining. TRACP activity varied in DCs from donor to donor but was reproducible on repeated examinations of each sample. Samples could be stained for simultaneous analysis of surface antigens and intracellular TRACP activity, provided certain technical details were observed. The TRACP reaction time should not exceed 9 min and the cell number should not exceed 2 × 105/100 μl test. Fluorescent surface labels did not affect the intensity of the TRACP stain, but the intensity of some surface labels may be diminished by elution of low-affinity antibodies during the TRACP reaction. Readjustment of the threshold settings in triple-labeled cells is needed to compensate for this phenomenon. Intracellular TRACP activity can be quantitated in subpopulations of cells within mixed cell populations by flow cytofluorometry using simple cytochemical methods in combination with fluorescent antibodies to cell-surface and other differentiation antigens. The cytochemical method should be useful for basic investigations of differentiation, maturation, and function of macrophages, DCs, and osteoclasts, and for diagnosis and management of hairy cell leukemia.
Keywords
T
We have used cytochemistry on cell smears and polyacrylamide gels to study TRACP expression in dendritic cells (DCs) derived from human blood monocytes (Janckila et al. 2002a). The purpose of the present study was to develop a cytochemical method for rapid quantitation of TRACP-expressing cells by flow cytometry in which dendritic cells were used as the model cell type. Results from this study show that the cytochemical method for TRACP enzymic activity can be used in combination with immunochemical staining of distinct surface membrane markers for flow cytometric analysis. Both the number of positive cells in the population and the level of TRACP activity in individual cells can be reproducibly quantitated.
Materials and Methods
Cell Sources
Human monocyte-derived phagocytic dendritic cells (DCs) were prepared as previously described (Romani et al. 1996) and used to optimize TRACP staining methods. Briefly, mononuclear cells (MNCs) were prepared by Ficoll-Hypaque gradient centrifugation of buffy coat cells from normal human donor blood obtained from the hospital blood center. After washing MNCs twice in Hanks's balanced salt solution (HBSS), the cells were suspended to 3-5 × 106/ml in AIM V medium (Gibco BRL, Gaithersburg, MD) supplemented with 1% fetal bovine serum and seeded into 100-mm tissue culture dishes (7 ml/dish). Monocytes were allowed to adhere for 2 hr at 37C, after which the non-adherent cells were decanted and the plates rinsed gently three times each with 5 ml HBSS. DC cultures were initiated by adding 6 ml of AIM V medium supplemented with 1% fetal bovine serum, 20 ng/ ml interleukin-4 (Leinco Technologies; St Louis, MO) and 50 ng/mL GM-CSF (Leinco Technologies) to each dish. On the third day of culture, cells were fed with an additional 3 ml of fully supplemented AIM V medium containing IL-4 and GM-CSF. Dendritic cells were harvested on day 6 by gentle pipetting to remove loosely adherent cells. Three to five ml HBSS containing 2 mM ethylene diamine tetraacetate (EDTA) was added to each plate and incubated for 15-30 min at 37C to release the strongly adherent cells, which were pooled with the loosely adherent cell harvest. DCs were pelleted by centrifugation at 400 × g for 10 min and washed two times in HBSS. For convenience and for repeated studies, washed DCs were cryopreserved at 2-10 × 106/ml in 90% fetal bovine serum/10% dimethyl sulfoxide (DMSO) until used. After thawing, cell viability was greater than 85% in all cases. There is the possibility that TRACP expression characteristics could change within a sample due to freeze–thaw artifacts, but we did not investigate this possibility. All of our samples were treated similarly to provide consistency within the study.
Cytochemistry of TRACP on Cell Smears
Cytocentrifuged smears were prepared from day 6 DCs and stained for TRACP activity using published methods (Yam et al. 1987) employing naphthol-ASBI phosphate (Sigma Chemical St Louis, MO) as substrate and fast red violet LB salt as coupler (Sigma). The cytochemical stain reaction was stopped after 9 min to provide light microscopic validation for flow cytoenzymology results.
Cell Extraction and Immunoassay
In four experiments, DCs were harvested for immunoassay of intracellular TRACP activity and protein. Washed DCs were counted, pelleted, and resuspended to 107 cells /ml in lysis buffer (10 mM Tris/300 mM NaCl/0.5% NP-40/1 mM EGTA, pH 7.5) containing 1 mM phenyl methylsulfonyl fluoride and 1 μg/ml each pepstatin and leupeptin. Lysates were placed on ice for 30 min, then frozen at –70C until analyzed. Lysates were thawed and centrifuged to remove insoluble material and the supernatant assayed for TRACP activity and protein by published methods (Nakasato et al. 1999). Anti-TRACP antibody 14G6 was used as capture antibody for both assays. Peroxidase-conjugated anti-TRACP J1B antibody was used as detection antibody in a two-site sandwich assay for TRACP protein. Two to five microliters of lysate was used for TRACP activity assay and results were expressed as mU/106 cells. One microliter of lysate was used for TRACP protein assay and results expressed as ng/106 cells. Dilutions of 4-nitrophenolate ranging from 10 U/l to 0.16 U/l were used as calibrators for TRACP activity assay. Dilutions of recombinant CHOTRAP (Janckila et al. 2002b) from 5-0.31 μg/liter were used as calibrators for TRACP protein assay.
Cytochemical TRACP Reaction for Cytofluorometry
Cryopreserved cells were rapidly thawed and washed twice in PBS. Afterward, the viability was always greater than 85%. Viable cells were placed in microcentrifuge tubes and pelleted. All subsequent incubations were carried out on ice. Cells were fixed for 1 min in a solution of 4% paraformaldehyde in PBS and then permeabilized by adding Triton X-100 to a final concentration of 0.02% and incubating for another 2 min. Fixed cells were washed by centrifugation once in PBS and once in TRACP stain buffer (50 mM MES, pH 6.3, containing 50 mM Na tartrate) and adjusted to 2 × 105 to 4 × 106 cells/ml. Aliquots of 50 μL cells in TRACP stain buffer were then prewarmed to 25C. A 2 × staining solution was prepared immediately before use by adding 80 μl stock naphthol-ASBI phosphate (12.5 mg/ml in dimethyl formamide) and 20 μl stock fast red violet (10 mg/ml in DMF) to 900 μl prewarmed MES/tartrate buffer. Both ASBI-P and FRV solutions were stored in single-use aliquots. To initiate the enzymatic reaction, 50 μl of prewarmed 2 × staining solution was added to the warmed cells and incubated for precisely 9 min. The reaction was stopped by adding 900 μl ice-cold PBS. To control for background fluorescence of cytochemical reaction product, ASBI-P substrate was omitted while fast red violet salt remained in the solution. For some experiments to optimize the method, the reaction time was varied from 3 to 15 min and the cell number varied from 2 × 105 to 2 × 107 cells/ml.
Simultaneous Staining of Surface Antigens and TRACP Activity
Anti-CD86-fluorescein isothiocyanate (FITC) and anti-CD11c-phycoerythrin (PE) were purchased from BD Biosciences (Singapore). To demonstrate surface membrane antigens together with cytoplasmic TRACP activity, surface antigens were labeled first, followed by TRACP staining. Fixed and permeabilized cells were suspended in 50 μl PBS with pre-tested optimal amounts of commercial fluorochrome-labeled monoclonal antibodies and incubated on ice for 30 min. After washing twice in PBS containing 2% BSA, the cells were subjected to TRACP cytochemical staining as described above. The triple-labeled cells were used immediately for cytofluorometric analysis.
Cytofluorometric Analysis
Cytofluorometry was performed with a Becton-Dickinson FACSCalibur system equipped with a 488-nm blue laser and a 635 red diode laser for multicolor fluorescence, in addition to forward scattering and side scattering measurements. FITC-labeled antibodies and PE-labeled antibodies were measured using FL1 at 530 nm and FL2 at 585 nm, respectively. TRACP activity revealed by naphthol-ASBI/FRV-LB reaction was detected in FL3 at 670 nm. All cytofluorometric data were subsequently analyzed and graphically displayed using B-D Cell Quest software.
Results
Cytofluorometry of TRACP Activity by Cytochemical Staining
Using standardized final concentrations of substrate (1 mM) and coupler (0.25 mM) for cytochemical staining of intracellular TRACP in fixed cells, we optimized the number of cells per 100 μl test and the reaction time to achieve a mean fluorescence index (MFI) of the DC population in the linear portion of the kinetic curve. This way, strong positive and weak positive cells could be differentiated. The reaction time to maximal staining intensity was approximately 15 min. The kinetics of fluorochrome production were linear from 3 to 9 min using 105 cells per test or less (Figure 1). When the cell number exceeded 5 × 105 per test the reaction kinetics deviated from linearity resulting in substantially lower fluorescence at 9 min due to substrate depletion. We also observed that the MFI for TRACP cytochemistry increased in a time-dependent manner when cells were stored at 4C in the fixed state before staining and analysis (data not shown). This was probably due to mild reduction of the di-iron center of TRACP or to proteolytic activation of TRACP by intracellular proteases. Proteolytic activation of TRACP has been reported previously for purified TRACP stored at 4C (Marshall et al. 1997). MFI also increased during storage of intact cells after TRACP staining (data not shown). We interpret this to be due to residual substrate present in the cells after staining and washing.
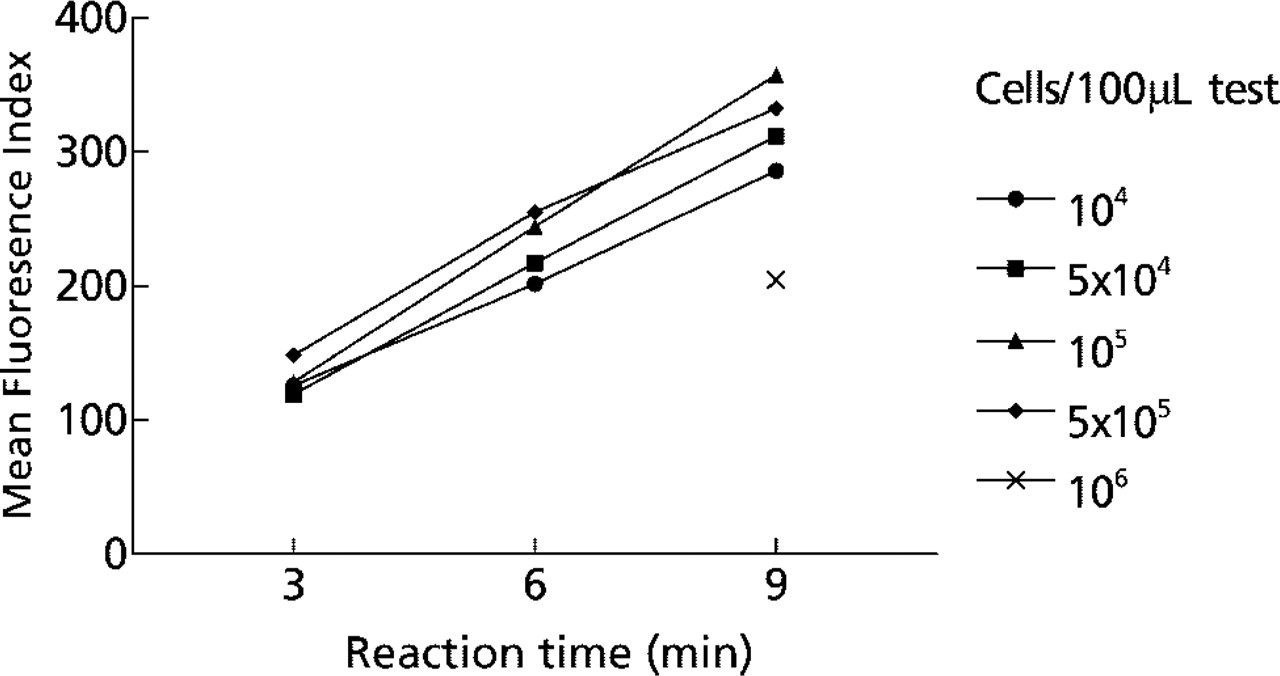
Flow cytometric analysis of TRACP enzyme activity in human monocyte-derived dendritic cells (DCs). Kinetics of mean fluorescence (FL 3) produced by naphthol ASBI-P/FRV-LB reaction at pH 6.35 and 25C using various numbers of cells from 104 to 106 cells/ 100 μl assay tube.
TRACP was expressed in greater than 90% of DCs from all eight cases tested. Although the intensity of TRACP expression varied from donor to donor, there was a high degree of reproducibility when the enzyme reaction was performed on freshly thawed cells and analyzed immediately (Table 1). We also observed that subpopulations of smaller DCs often expressed more TRACP activity than larger cells, as defined by forward scatter (data not shown). The case-by-case variation in TRACP expression was validated in DCs from four donors by light microscopy of stained DCs (Figure 2A) and by immunoassay of TRACP in cell extracts (Figure 2B). These findings suggest that flow cytofluorometry can be used for quantitative assessment of intracellular TRACP expression.
Co-expression of Cytoplasmic TRACP Activity and Surface Markers of Dendritic Cells
To use flow cytofluorometry of TRACP most effectively, it would be advantageous to simultaneously reveal surface markers with specific fluorescent antibodies. In Figure 3 are data from a representative experiment showing the results of this method. The gated population of DCs (Figure 3A) co-expressed CD86 and CD11c (Figure 3B), which are commonly used as markers for DCs. Background fluorescence caused by FRV coupler alone in control TRACP activity incubations is nil (Figures 3C and 3D) and did not affect detection of FITC-labeled CD86 or PE-labeled CD11c (Figures 3E and 3F). On staining TRACP activity with ASBI-P substrate, greater than 90% of DC were positive (Figures 3G and 3H), and the numbers of CD86-positive cells (Figure 3I) and CD11c-positive cells (Figure 3J) remained mostly unchanged. There were apparent correlations among CD86, CD11c, and TRACP expression by DCs from this donor. However, these relationships were not evident in DCs from all donors.
Reproducibility of cytoenzymology reaction for TRACPa
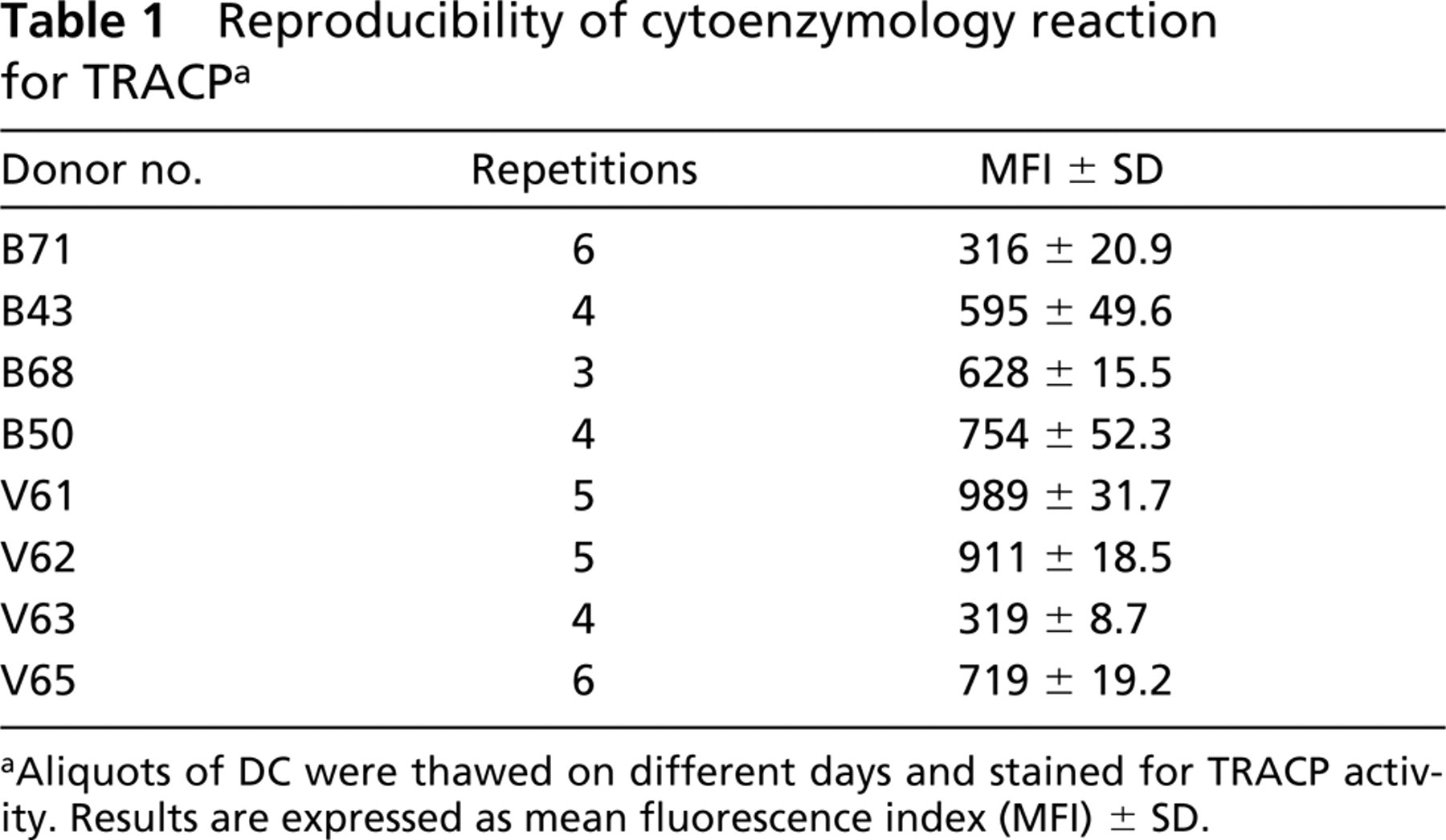
Aliquots of DC were thawed on different days and stained for TRACP activity. Results are expressed as mean fluorescence index (MFI) ± SD.
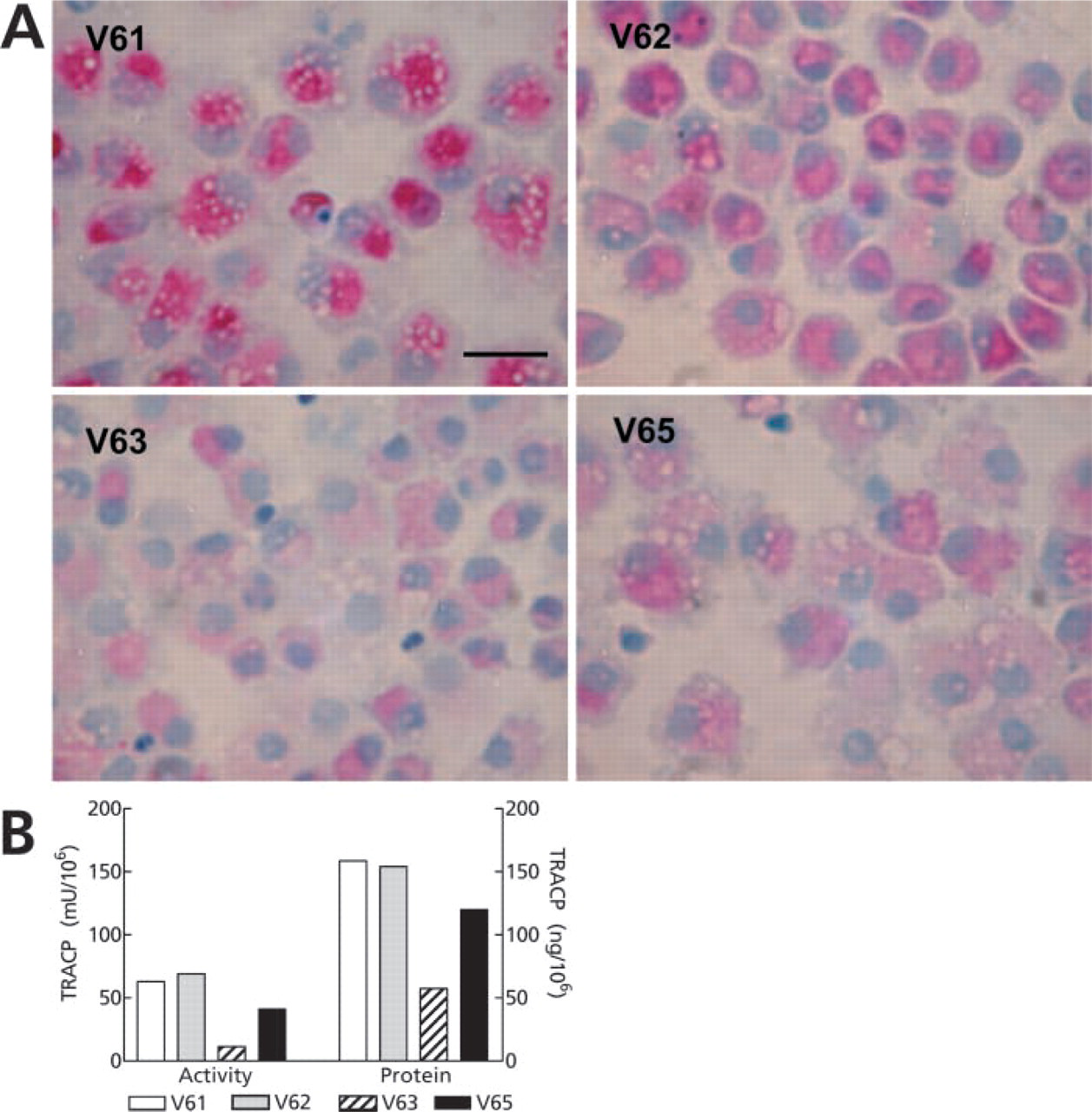
Cytocentrifuge smears of DCs from donors V61, V62, V63, and V65 stained for TRACP activity using naphthol-ASBI-P and FRV-LB. Bar = 20 μm. (
Sometimes we observed that the MFI of some surface marker stains was diminished after the TRACP cytochemical reaction compared to cells that were not stained for TRACP. Experiments (not shown) using antibodies from different sources and adjustments of TRACP reaction buffer concentration revealed that this was probably due to elution of lower-affinity antibodies during the TRACP reaction. The effect could be minimized, but not completely eliminated, by keeping the ionic strength of the TRACP reaction at approximately 50 mM. Therefore, to compare surface antigen staining before and after TRACP staining, the threshold settings were readjusted for analysis of triple-stained cells to partially correct for the elution effect. Table 2 summarizes quantitative data from cyto-fluorometric analysis of cell surface CD11c and CD86 expression before and after TRACP staining in preparations from five normal donors repeated on two to five separate occasions. CD11c and CD86, like TRACP, varied from case to case, were slightly reduced after TRACP cytochemical staining, but were reproducible. Table 3 summarizes data showing that surface labeling of different fluorescent antibody pairs had no effect on the intensity of detectable TRACP expression.
Discussion
Cytofluorometry is a powerful technology that allows precise quantitative description of complex cell mixtures. With it, it is now possible to define the type and differentiation stage of neoplastic cells and offer more accurate diagnoses of leukemias, lymphomas, and other cancers (Leith and Willman 2001). Currently, such phenotypic analyses rely heavily on fluorochrome-labeled antibodies reactive to surface and cytoplasmic antigens of defined lineage specificity. Before the advent of monoclonal antibodies, cytochemistry of intracellular enzymes was used with success for the same cell characterization purpose (Scott 1989; Li et al. 1996), particularly in the classification of acute leukemias. Early attempts to quantitate the reactions by flow cytometry met with partial success (Ansley and Ornstein 1971), but the methods have not been routinely implemented due to limited availability of suitable substrates and cell specificity of the enzyme reactions. Naphthol-ASBI phosphate is a useful substrate for cytochemical staining of TRACP activity in cells and tissues (Li et al. 1970). This substrate is fluorescent and can be used for biochemical determination of acid and alkaline phosphatases as well (Vaughan et al. 1971). We have recently shown it to be a relatively specific substrate for osteoclastic TRACP isoform 5b and therefore of potential use for selective biochemical assay of osteoclastic TRACP activity in serum (Janckila et al. 2001). The enzymatic reaction product, naphthol-ASBI, reacts readily with the diazonium salt fast red violet-LB (“simultaneous capture” principle) to form a highly chromogenic insoluble complex. This complex remains fluorogenic emitting at 670 nm, so it can be measured in FL3 of cytofluorometer. The fluorescence characteristics of N-ASBI/FRV-LB thus allow quantitative analysis of TRACP activity in cell populations and individual cells. Other chromogenic substrates and diazonium salts have been used successfully for cytochemical staining of TRACP on smears and tissues (Janckila et al. 1978). We have not yet tested their effectiveness for cytofluorometry, which will depend largely on their spectral characteristics and stability.
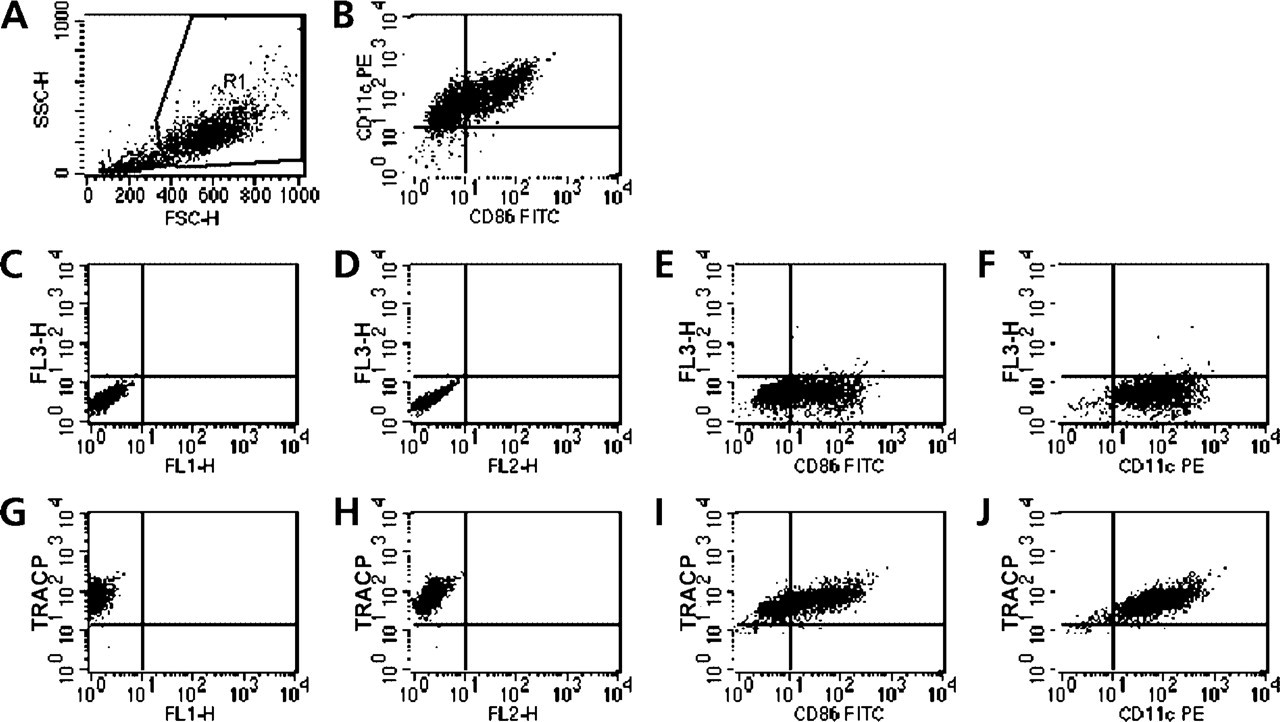
Cytofluorographic analysis of day 6 DCs from donor B71. The gated population of DCs (
Effect of TRACP cytochemical reaction on surface marker expressiona
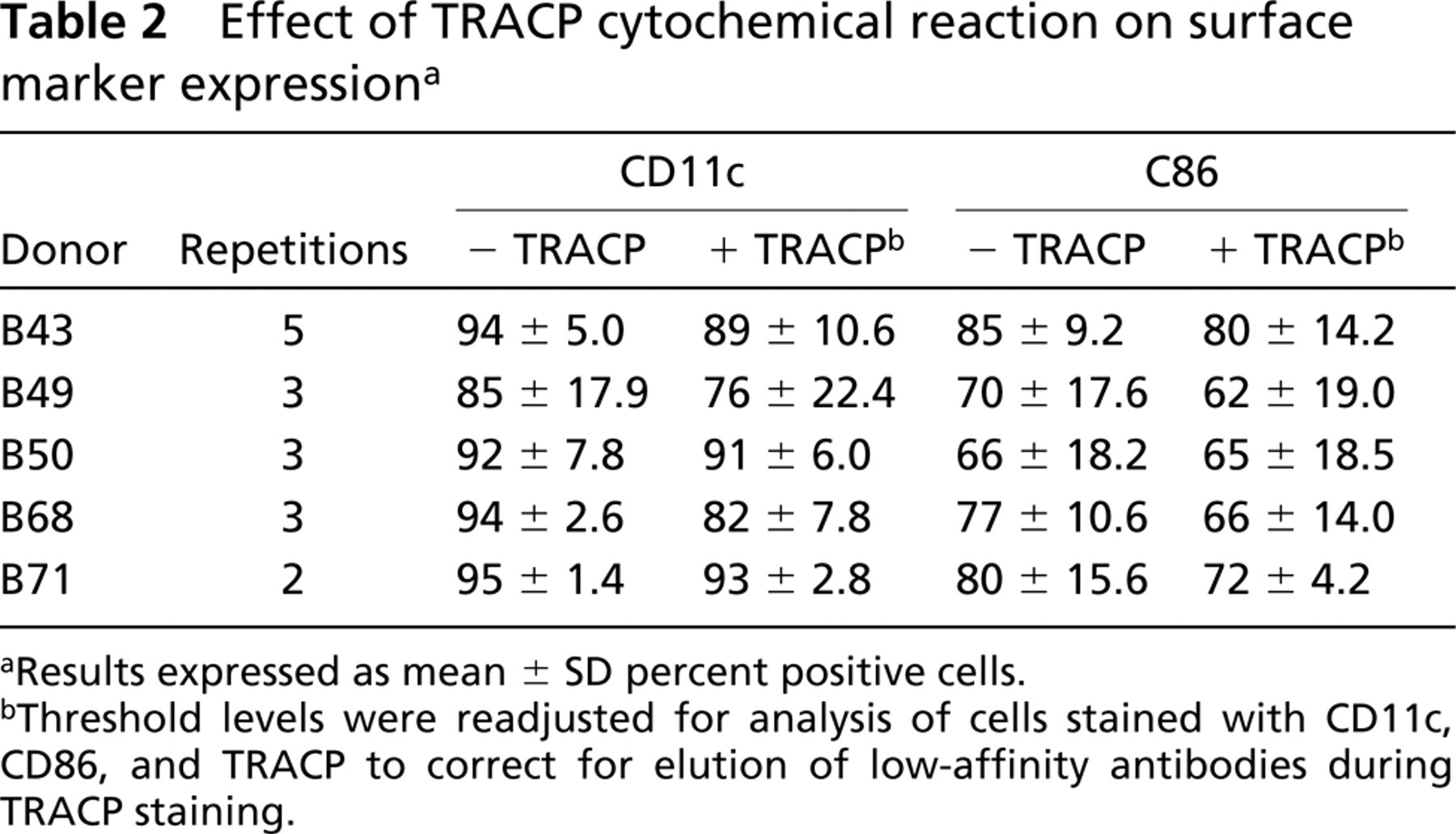
Results expressed as mean ± SD percent positive cells.
Threshold levels were readjusted for analysis of cells stained with CD11c, CD86, and TRACP to correct for elution of low-affinity antibodies during TRACP staining.
Effect of surface antigen staining on TRACP staining intensitya
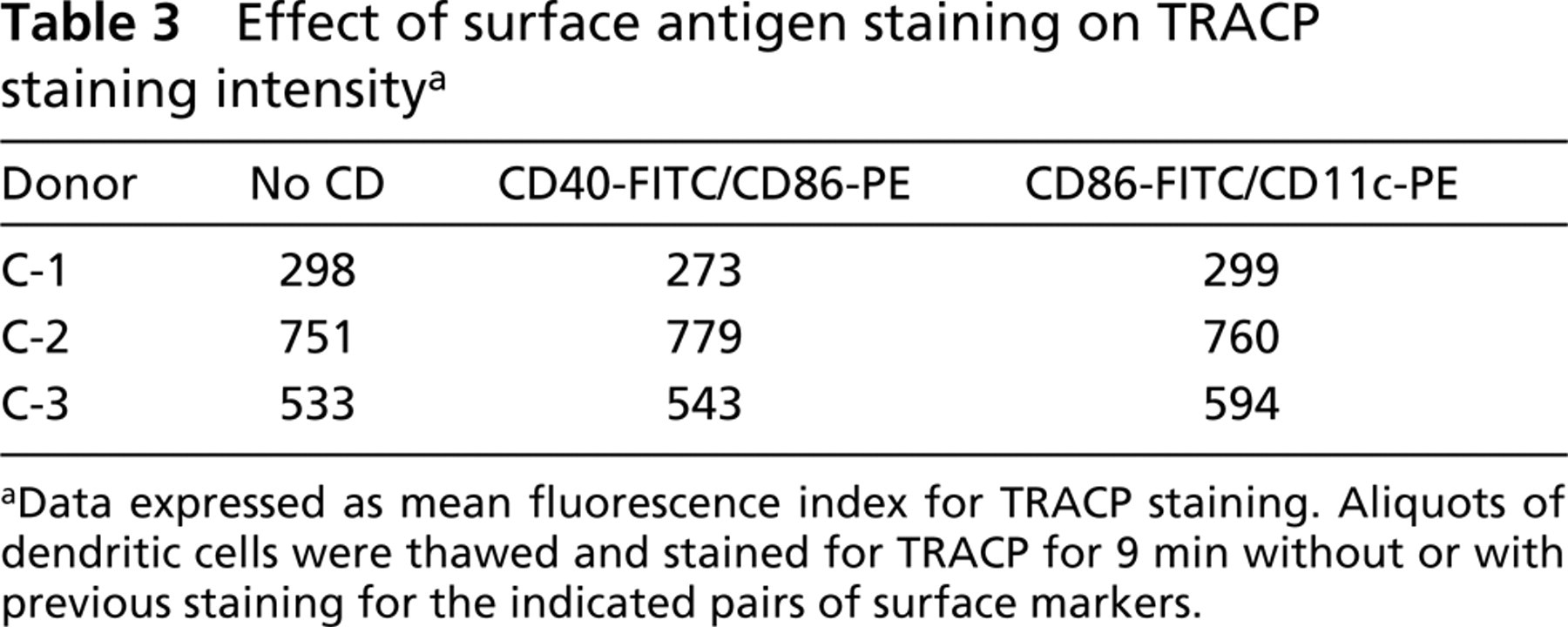
Data expressed as mean fluorescence index for TRACP staining. Aliquots of dendritic cells were thawed and stained for TRACP for 9 min without or with previous staining for the indicated pairs of surface markers.
In these studies we established a cytochemical method for quantitation of intracellular TRACP activity in cell suspensions via flow cytoenzymology. The method can be performed alone or in combination with immunolabeling of cell surface antigens to demonstrate TRACP expression in phenotypically defined cell populations. The availability of these novel methods to demonstrate TRACP activity would be of considerable practical and scientific value.
In practice, TRACP enzyme cytofluorometry is easy to perform. However, certain technical details require careful attention. The reaction is rapid with linear kinetics, reaching maximal intensity in approximately 15 min at pH 6.3 and 25C. By incubating for 9 min (within the linear range), the method can discriminate cells with high activity from those with low activity. Because the reaction is so rapid and sensitive, careful control of the incubation time and temperature is essential for reproducible results. The number of cells per assay should be kept at or below 2 × 105/100 μl sample. Beyond this, the reaction becomes non-linear, with diminishing fluorescence due to substrate depletion. Therefore, for consistent results, the concentration of cells in the test sample must be determined beforehand and adjusted to within a defined range. Finally, samples should be stained and analyzed on the same day they are prepared. TRACP activity in DCs increased significantly on refrigerated storage in the fixed state. Pure recombinant TRACP has been observed to undergo limited proteolytic cleavage of the “loop” peptide on storage to cause a marked rise in specific activity (Marshall et al. 1997). It is conceivable that TRACP in intact cells, stored in the refrigerator, may undergo similar limited proteolysis to cause a significant increase in MFI compared to freshly thawed cells.
Using this cytochemical technique under well-controlled conditions, the TRACP activity within individual DCs still varied considerably among different normal donors, although the percentage of positive cells was consistently greater than 90%. Most DCs (85-95%) from five donors tested strongly expressed CD11c. CD86 was variably expressed to a lesser degree (66-85%). We observed that some antibodies were significantly desorbed from cells after subsequent cytochemical staining for TRACP. We could minimize this effect by maintaining the ionic strength of the TRACP reaction buffer at approximately 50 mM. Even with careful readjustment of the threshold settings in the triple-labeled preparation, there was a measurable decline in surface marker fluorescence intensity in some instances. On the other hand, TRACP enzymatic activity was unaffected by prior labeling of cell surface antigens with fluorescent antibodies. To provide consistency throughout this study, all experiments were performed on cells that were fixed and permeabilized before any labeling reactions. This eliminated the problem of differential light scattering by fixed and unfixed cells. We realize that fixation may crosslink or denature some important epitopes on some target antigens recognized by monoclonal antibodies. It must be determined empirically, by each laboratory, which surface antigens are stable to fixation and which are not. For this method to work with labile antigens, the surface antigens must be labeled first, followed by fixation, permeabilization, and TRACP staining.
From a scientific point of view, cytofluorometry of intracellular TRACP will allow study of the temporal expression and possible functional relationships among TRACP and other defining markers of the various macrophage lineages as the cells grow, differentiate, and mature. The present method is a relatively simple one that allows the measurement of TRACP enzymatic activity at a defined endpoint, together with other cell surface antigenic markers. Therefore, it cannot make kinetic measurements of enzymatic activity in individual cells by repeated or continuous monitoring of the reaction, as can laser scanning cytometry (Bedner et al. 1998). Osteoclasts (OCs) are bone marrow-derived cells whose differentiation and maturation are under the influence of local cytokines and interactions with microenvironmental factors. Quantitative flow cytometry of TRACP expression by OCs in conjunction with other OC differentiation markers, such as calcitonin receptors, will facilitate the identification of OC precursors and the study of their differentiation and maturation. DCs are potent antigen-processing and -presentation cells (Banchereau and Steinman 1998), with TRACP expressed abundantly at a time when antigen processing efficiency is maximal. Indeed, it has been suggested that TRACP is directly involved in the antigen processing function itself in DC and macrophages (Hayman et al. 2000; Bune et al. 2001; Raisanen et al. 2001). Quantitative assessment of intracellular TRACP activity in conjunction with surface differentiation and endosomal compartment markers will aid investigations of the potential role of TRACP in antigen processing by DCs and other antigen-presenting cells. Cytochemical and immunocytochemical demonstration of TRACP in smears and tissue sections has been used for diagnosis of hairy cell leukemia (Hoyer et al. 1997). These novel cytofluorometric methods for intracellular TRACP, particularly in combination with other surface markers, could make diagnosis and evaluation of residual disease in HCL more quantitative and accurate. The clinical sensitivity and specificity of cytofluorometry of intracellular TRACP in HCL merit further study.
Footnotes
Acknowledgements
Supported by a grant from the Research Service of the US Department of Veterans Affairs (AJJ) and a grant (DOH91-TD.1001) from the Department of Health, Taiwan, ROC (WKY).
We thank Ms Luann Jaggers for her technical assistance. We gratefully acknowledge Dr Jussi Halleen for his generous gift of anti-TRACP J1B antibody.