
Abstract
Tartrate-resistant acid phosphatase (TRAP) is a histochemical marker of the osteoclast. It is also characteristic of monohistiocytes, particularly alveolar macrophages, and is associated with diverse pathological conditions, including hairy cell leukemia and AIDS encephalopathy. To study the biology of this enzyme, we investigated its expression and activity in mouse tissues. Confocal fluorescence studies showed that TRAP is localized to the lysosomal compartment of macrophages. In adult mice, high activities of the enzyme were demonstrated in bone, spleen, liver, thymus, and colon, with lower amounts in lung, stomach, skin, brain, and kidney. Trace amounts were detected in testis, muscle, and heart. Expression of TRAP mRNA was investigated in tissue sections by in situ hybridization and protein expression was monitored by histochemical staining or immunohistochemically. TRAP is widely expressed in many tissues, where it is associated with cells principally originating from the bone marrow, including those of osteoclast/macrophage lineage. The cellular distribution of TRAP mRNA and enzyme antigen in the tissues corresponds closely to that of cells staining with an antibody directed to the CD80 (B7) antigen. Therefore, to confirm its putative localization in dendritic cells, isolated bone marrow dendritic cells were matured in culture. These co-stained strongly for TRAP protein and the CD80 antigen. These studies demonstrate that TRAP is a lysosomal enzyme that is found in diverse murine tissues, where it is expressed in dendritic cells as well as osteoclasts and macrophages, as previously shown.
M
Although the human gene maps to chromosome 19 and contains a putative N-terminal lysosomal leader sequence (Lord et al. 1990), the cognate protein is secreted into the resorptive vacuole beneath the ruffled border of the osteoclast during bone resorption (Reinholt et al. 1990). Lately, evidence has been presented that TRAP is not secreted into the ruffled border but rather is present in vesicles that are fused to transcytotic vesicles. These vesicles transport matrix degradation products from the ruffled border to a specialized membrane area in the basolateral membrane where the enzyme is secreted together with the matrix degradation products (Halleen et al. 1999). It has been proposed that TRAP participates in specialized electrochemical reactions associated with resorption of the bone matrix by the osteoclast (Hayman and Cox 1994) and is capable of forming hydroxyl radicals (Sibille et al. 1987). Macrophages from TRAP knockout mice show an altered production of free radicals (unpublished results).
TRAP activity can be detected in the serum of mammals, where it exists as a complex with α2-macroglobulin (Shaffer-Brehme et al. 1999). The activity of the enzyme is pathologically increased in conditions where bone resorption is enhanced, including Paget's disease and hyperparathyroidism (Lau et al. 1987; Chamberlain et al. 1995). Expression of TRAP is associated with the activation and differentiation of osteoclasts, macrophages, and other cells of monohistiocytic phenotype, such as the Gaucher cell. Gene expression has been demonstrated in cells originating from granulocyte-monocyte progenitors, osteoclasts, and in alveolar and monocyte-derived macrophages, but was undetected in human blood monocyte or normal splenic tissue (Bevilaqua et al. 1991). Hairy cell transformation of B-lymphocytic leukemia cells is also associated with unexplained expression of TRAP (Mende et al. 1975).
Hitherto, TRAP has been considered to be expressed selectively in adult tissues, e.g., pulmonary alveolar macrophages in the lung (Efstratiadis and Moss 1985), and in the infiltrating macrophages of the brains of patients suffering from HIV-induced encephalopathy (Schindelmeiser et al. 1989). Because TRAP occurs in cells of macrophage origin as well as in serum, key questions are raised as to its origin and distribution. Accordingly, we aimed to determine the constitutive expression of TRAP at the level of its transcript and as enzymatically active protein in parenchymal mammalian organs, including the brain.
Materials and Methods
Preparation of Monocyte-derived Macrophages
Heparinized human blood diluted 1:1 with Hank's balanced salt solution was underlayered with lymphocyte separation medium (Organon Teknika; Cambridge, UK), and centrifuged at 1800 rpm for 30 min at room temperature (RT). The lymphocyte layer was aspirated, diluted with Hank's balanced salt solution (Sigma; Dorset, UK), and spun at 1400 rpm for 10 min at RT. The pellet was washed in RPMI 1640/10% FCS (Sigma), centrifuged at 1200 rpm for 5 min, and resuspended in the same medium. The cell suspension was added to fibronectin-coated glass coverslips, incubated for 45 min at 37C, and washed six times with RPMI/10% FCS to remove nonadherent cells. Cells were differentiated for 72 hr in RPMI supplemented with 10% FCS, 2 mM
Dendritic Cell Isolation
Dendritic cell isolation was based on a method by Inaba et al. (1992). Bone marrow was extracted from the femurs of male mice aged 6-8 weeks. Cells were plated at 1 million/ml in RPMI 1604 in the presence of 10% FCS (First Link; West Midlands, UK) 2 mM glutamine, 20 μg/ml gentamicin, 100 U/ml penicillin, 100 μg/ml streptomycin, 10 mM HEPES (all from Sigma), and 20 ng/ml GM-CSF (First Link). Medium was changed every 2 days for 6 days. Semiadherent aggregates of dendritic cells were transferred to fresh dishes and incubated at 37C for 24-48 hr. Mature dendritic cells that were nonproliferative and nonadherent were collected by gently swirling the dish.
Immunofluorescence Microscopy of Human Macrophages
For immunostaining, macrophages were incubated with primary antibodies in PBS/1% BSA (Sigma) for 45 min. These were rabbit polyclonal uteroferrin or mouse IgG monoclonal to the lysosomal marker LAMP-1 (lysosomal associated membrane marker 1; a gift of Dr. Jordan Pober, Yale University). After washing three times with PBS/1% BSA, macrophages were incubated with secondary fluorescein isothiocyanate-conjugated (FITC-conjugated; Sigma) or rhodamine-conjugated (Dako; Bucks, UK) antibodies for 45 min. Macrophages were washed twice with PBS/1% BSA and once with PBS, and coverslips were mounted on glass slides in a mixture of glycerol and PBS (9.1 v/v) containing 0.1% p-phenylenediamine. Cells were examined with a BIORAD MRC 600 confocal microscope (Hemel Hempstead, UK).
Immunofluorescence Microscopy of Murine Dendritic Cells
Dendritic cells were air-dried onto slides and fixed in methanol/acetone (1:1) at 20C for 15 min. Cells were washed three times with PBS, permeabilized with 0.1% Triton X-100 for 5 min, and incubated with goat antibody directed to the CD80 (B7) antigen (Santa Cruz Technology; Santa Cruz, CA) and rabbit antibody to porcine uteroferrin (Echetebu et al. 1987) overnight at 4C. Control sections were incubated with nonimmune purified sheep and rabbit IgG but otherwise with a complete set of reagents including secondary antibodies. Slides were washed three times with PBS and incubated at RT with FITC-conjugated and rhodamine-conjugated secondary antibodies (Chemicon; Harrow, UK). Cells were washed three times with PBS and mounted with AF1 (Citifluor; UKC, Canterbury, UK). Cells were examined with a laser BIORAD MRC 1000 microscope.
Assay of TRAP Activity in Murine Tissues
Tissue samples of liver, spleen, kidney, heart, lung, brain, thymus, skin, bone, testis, colon, stomach, and muscle were frozen in liquid nitrogen immediately after death. The tissues (100 mg) were homogenized in 0.5 ml 0.4 M sodium acetate, pH 5.6, containing 1% w/v Triton X-100. To assay specifically for TRAP (band 5), immunoabsorption with immobilized rabbit antibodies to porcine uteroferrin was carried out (Echetebu et al. 1987) after neutralizing 13,000 times; g supernatant tissue extracts to pH 7.5 with Tris base. For controls, immobilized protein A was added to the extract in place of the antibody. TRAP activity was finally determined spectrophotometrically before and after immunoabsorption in the presence of 0.1 M L(+) sodium tartrate at pH 5.6 using 10 mM 4-nitrophenyl phosphate as substrate (Hayman et al. 1989). Control assays were also performed on tissue extracts from mice lacking this phosphatase isozyme as a result of targeted gene disruption.
Localization of TRAP in human macrophages. Isolated macrophages (
Northern Blotting
Multiple-choice Northern blots were purchased from Cambridge Biosciences (Cambridge, UK). Blots were made with 2 μg poly A+ RNA per lane from murine spleen, liver, lung, testis, skin, brain, heart, kidney, stomach, small intestine, and skeletal muscle aged 9-10 months and thymus aged 8-12 weeks. The membranes were wetted in 4 times; SSC for 20 min and incubated for 2 hr at 65C in Rapid-hyb buffer (Amersham; Poole, UK). Human TRAP cDNA (25 ng) was labeled using the Prime-a-Gene Labeling System from Promega (Southampton, UK). The probe was purified down a column of Sephadex G-50 and added to the prehybridization solution with the blot. Hybridization was carried out for 2 hr at 65C. Blots were washed at the same temperature: three times for 5 min with 2 times; SSC/0.1% SDS and subsequently with 2 times; 0.25 times; SSC/0.1% SDS for 30 min. Blots were exposed to Hyperfilm TM-βmax (Amersham) at −80C for 4 days.
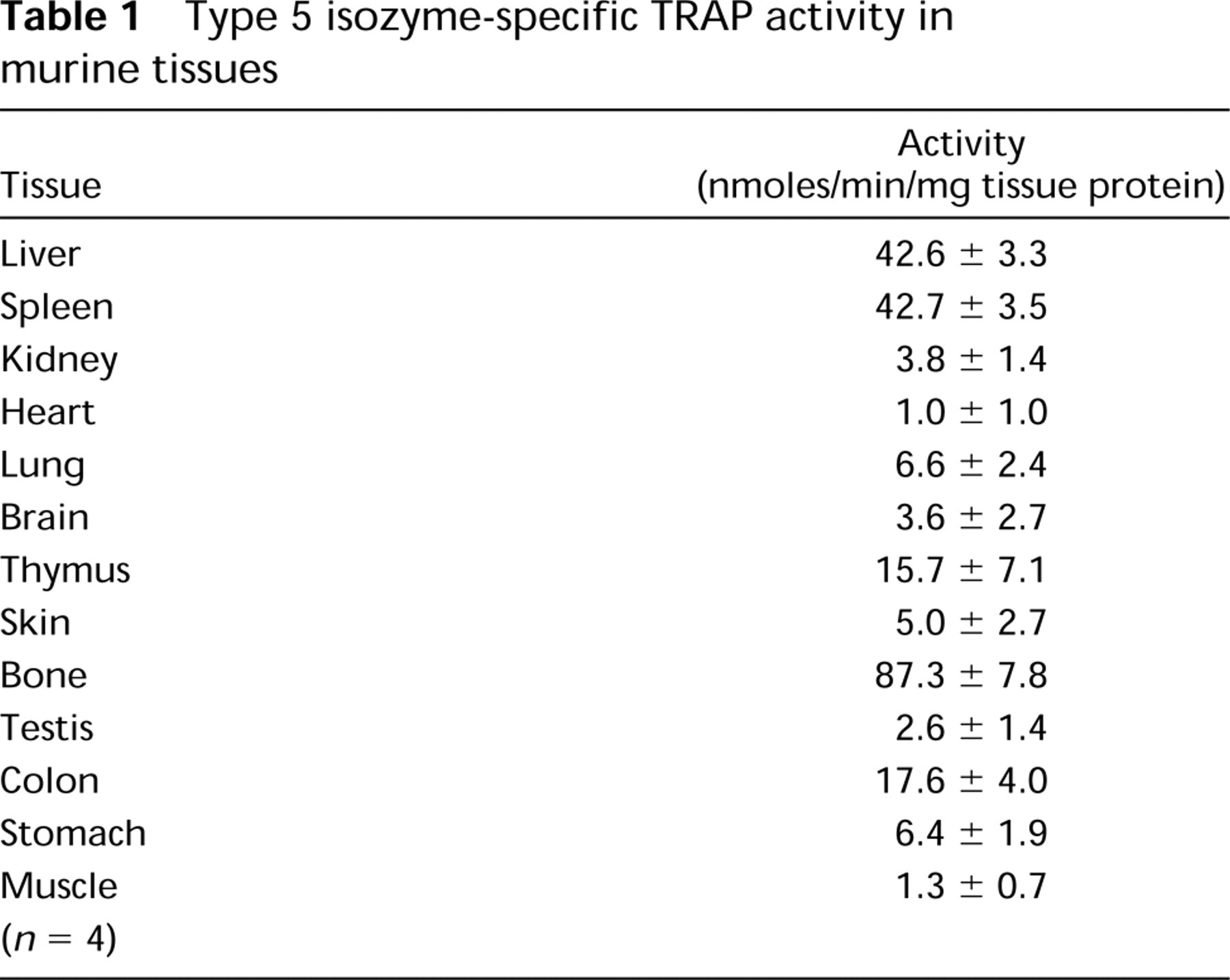
Type 5 isozyme-specific TRAP activity in murine tissues
Tissue Preparation for Histology and In Situ Hybridization Studies
Bones were removed immediately after death and dissected free of muscle. The individual bones were dipped in 5% polyvinyl alcohol and frozen in a hexane bath. All other tissues—spleen, liver, lung, heart, kidney, skin, and brain—were snapfrozen in liquid nitrogen. Frozen material was kept at −70C until cut into sections (10-mm) on a Bright Microtome 5030 (Huntingdon, UK).
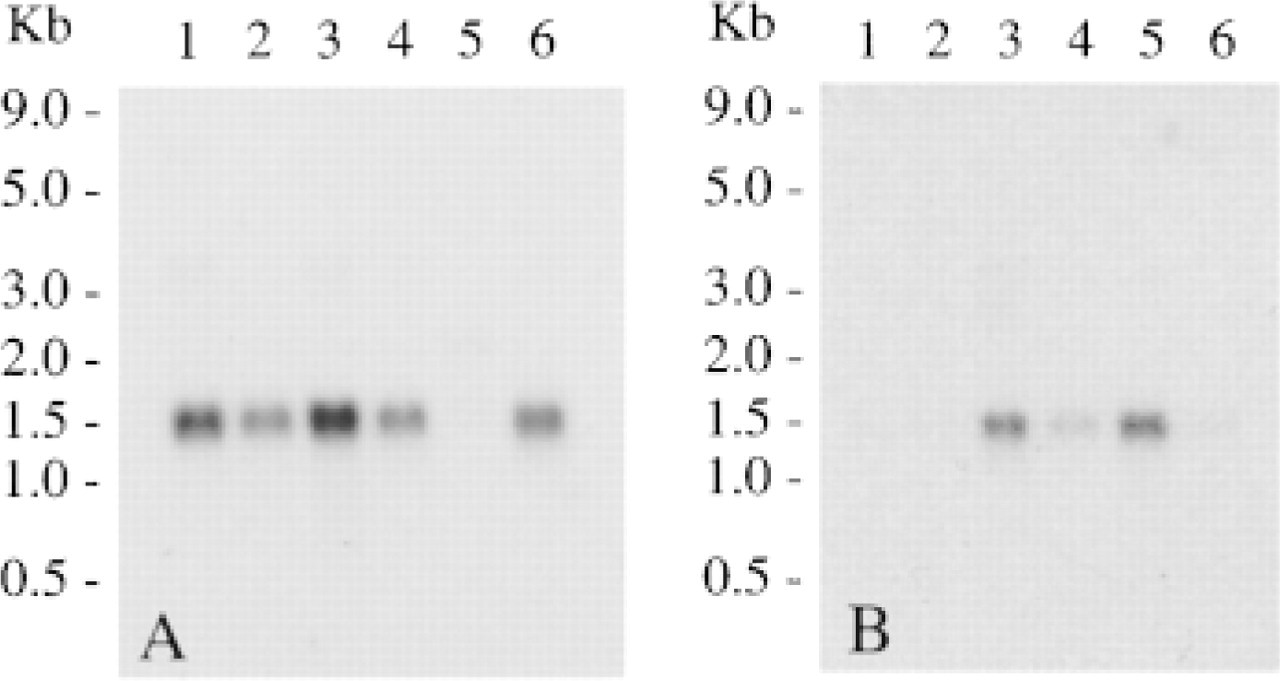
TRAP expression in mouse tissues. Northern hybridization analysis of mouse poly A+ RNA with a radiolabeled human TRAP cDNA showing positive transcripts of 1.5 kb. Lane A1, spleen; Lane A2, thymus; Lane A3, liver; Lane A4, lung; Lane A5, testis; Lane A6, skin; Lane B1, brain; Lane B2, heart; Lane B3, kidney; Lane B4, stomach; Lane B5, small intestine; Lane B6, skeletal muscle.
Histochemical Staining for TRAP
Unfixed, undecalcified cryostat sections were stained for TRAP activity using the standard naphthol AS-BI phosphate postcoupling method, using Fast Red as the coupler, where naphthol phosphate cleavage gives rise to a red precipitate of a diazonium product. The incubation was carried out for 10 min at RT in 0.4 M sodium acetate buffer, pH 5.6, containing 2 mM naphthol AS-BI phosphate and 100 mM sodium tartrate. The reaction was stopped in distilled water and post-coupled in the same buffer containing 2 mM Fast Red for 2 min or until color developed, followed by washing in distilled water (Burstone 1958). Sections of tissues from TRAP knockout mice (Hayman et al. 1996) were used as controls.
Immunohistochemistry
Frozen sections were fixed in acetone at −20C for 20 min. After washing in PBS for 5 min, sections were incubated for 20 min in diluted normal blocking serum prepared from the species in which the secondary antibody was made. Excess serum was blotted from sections and primary antibody diluted in PBS was applied. The primary antibodies used were polyclonals: rabbit anti-uteroferrin and a goat anti-B7 (V17; Santa Cruz) antibody recognizing the dendritic cell marker CD80. Nonimmune serum was used for controls. After an overnight incubation at 4C, the slides were washed twice in PBS. Diluted biotinylated secondary antibody was applied for 30 min, followed by washing in PBS as before. An avidin and biotinylated horseradish peroxidase macromolecular complex (Vector Laboratories; Peterborough, UK), based on the methods of Hsu et al. (1981a,b) was added to the sections and left for 30 min. After washing in PBS, hydrogen peroxide and diaminobenzidine tetrahydrochloride (DAB) were applied to the sections and incubated at RT until the desired intensity developed. The slides were rinsed in tapwater, counterstained with hematoxylin, and mounted.
Probe Preparation
The human TRAP cDNA, 1359 bp isolated from human spleen (Lord et al. 1990) was cloned into the Eco RI site of pGEM-7Zf(+). 35S-labeled riboprobes were prepared from linearized templates which were transcribed from T7 or SP6 promotors of the pGEM vector to generate antisense and sense probes. The riboprobes were synthesized using the Promega in vitro transcription kit with [35S]-uridine 5′ (α-thio) triphosphate (New England Nuclear; Hounslow, UK). After transcription, cDNA templates were digested with RNase-free DNase I, extracted with phenol/chloroform, and unincorporated nucleotides were removed by centrifugation through Sephadex G-50 columns. Riboprobes were quantified in a Packard 1500 liquid scintillation analyzer (Canberra-Packard; Pangbourne, UK).
In Situ Hybridization
Cryostat sections of mouse tissues were placed on Polysine slides (Scientific Laboratory Supplies; Nottingham, UK) and analyzed for TRAP expression by in situ hybridization. Briefly, sections were fixed in 4% paraformaldehyde for 30 min, washed in PBS, and treated with proteinase K (1 μg/ml). Sections were acetylated with freshly prepared 0.25% acetic anhydride in 0.1 M triethanolamine-HCl, pH 8, for 10 min at RT. The sections were dehydrated in 70% ethanol, air-dried, and used for hybridization. Sections were prehybridized in hybridization solution without probe (50% formamide, 0.3 M NaCl, 20 mM Tris-HCl, pH 8.0, 5 mM EDTA, 1 times; Denhardt's solution, 10% dextran sulfate, 10 mM DTT, 0.5 mg/ml tRNA, denatured salmon sperm DNA, 50 μg/ml, added immediately before) in a humid environment of 50% formamide at 50C for 2 hr. Tissue paper coverslips were used during the prehybridization stage and were removed at the end of the incubation time, after which hybridization solution containing 1.5 times; 106 cpm of probe/100 μl/slide was applied, followed by a glass coverslip. Sections were hybridized for 18 hr at 50C. After hybridization, the slides were immersed in 50% formamide in 2 times; SSC for 1 hr at 50C to remove coverslips. Unhybridized probe was digested with RNase A (20 μg/ml) in 2 times; SSC at 37C for 20 min. The slides were then washed for 1 hr at 55C in 2 times; SSC, followed by a further hour in 0.1 times; SSC. Sections were dehydrated in 70% ethanol and air-dried. Slides were exposed to Hyperfilm TM-βmax (Amersham) for 5 days. Slides were dipped in K5 emulsion (Ilford; Cheshire, UK) and exposed for 1-2 weeks at 4C depending on the signal intensity on the X-ray film, and then developed in Kodak D-19 developer (KP Professionals; Cambridge, UK). Sections were counterstained with hematoxylin and eosin and mounted. Hybridization signals were observed and photographed using a Nikon Optiphot 2 microscope (Surrey, UK) under darkfield illumination. As a negative control, specimens were incubated with hybridization buffer containing a radiolabeled riboprobe harboring the sense sequence of TRAP.
Results
In human monocyte-derived macrophages, TRAP was exclusively localized to the lysosome (Figure 1). Immunostaining with FITC-labeled antisera to TRAP revealed a punctate immunofluorescence which colocalized with rhodamine-labeled antisera to the lysosomal membrane marker LAMP-1.
Protein extracts prepared from adult mouse tissues were assayed using a specific immunoassay for TRAP. The results shown in Table 1 revealed that TRAP activity was highest in bone, spleen, liver, thymus, and colon. Lower amounts of activity were measured in lung, stomach, skin, brain, and kidney. Traces of activity were measured in heart, testis, and muscle. Type 5 TRAP activity was undetectable in extracts from the genetically engineered mice lacking TRAP.
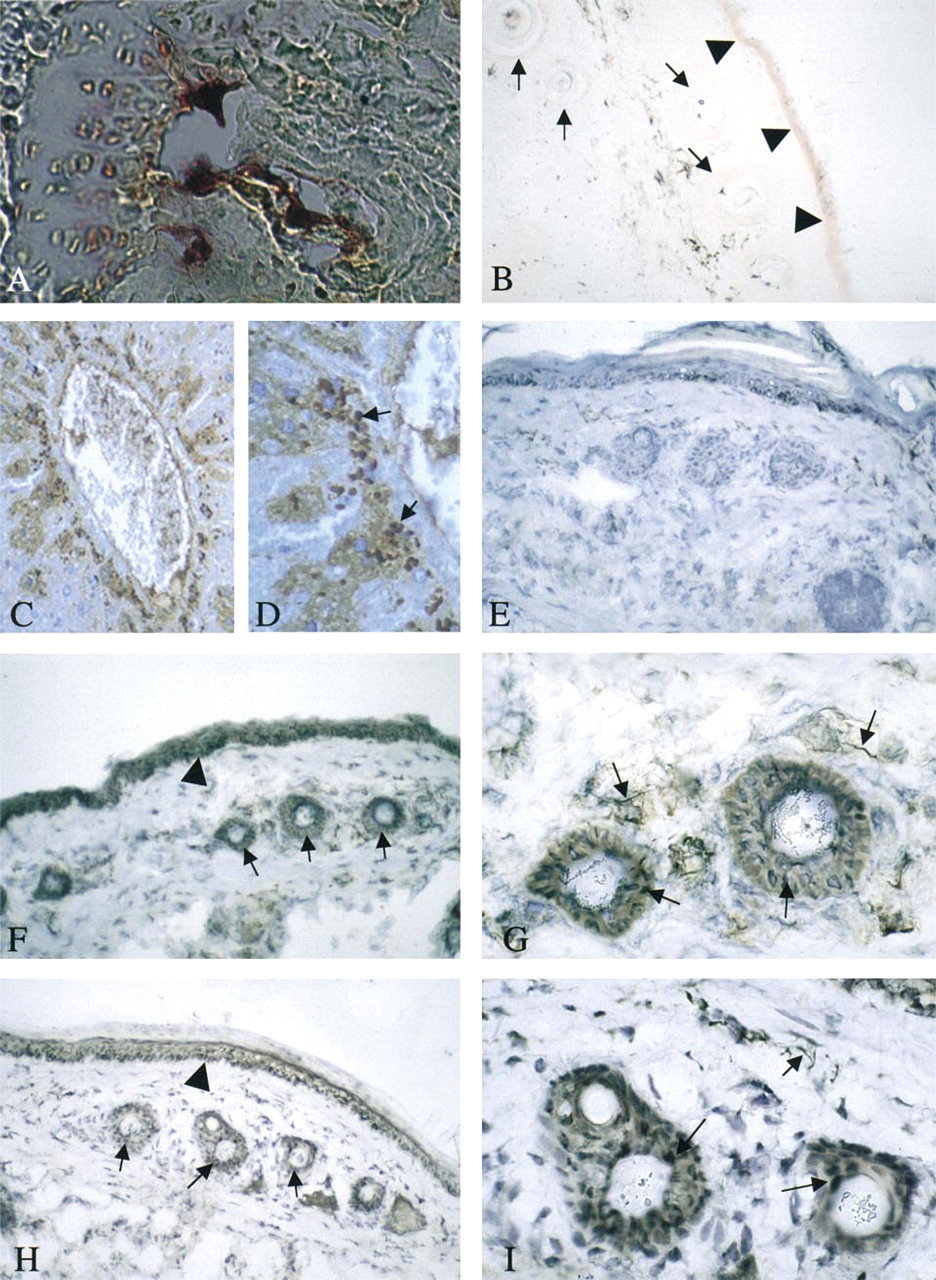
Histochemistry of TRAP in murine tissues. TRAP activity in osteoclasts in a section through the proximal end of a femur (
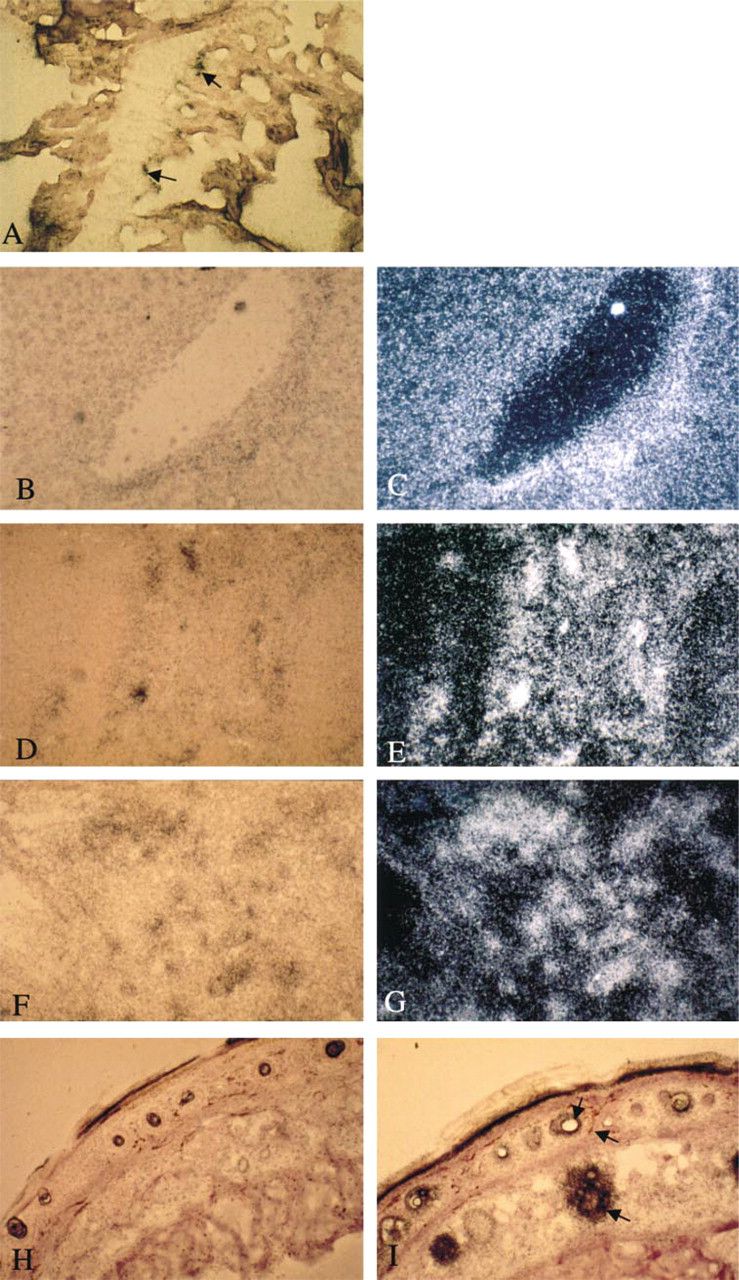
TRAP mRNA expression. Autoradiography of murine tissues hybridized with the 35S-labeled TRAP antisense riboprobe and counter-stained with hematoxylin. The figures represent lightfield (
An investigation of TRAP mRNA expression in 12 murine tissues was carried out by Northern blot analysis (Figure 2). Abundant expression of TRAP transcripts of the expected size of 1.5 kb were identified after hybridization of radiolabeled full-length human TRAP cDNA in spleen, thymus, liver, lung, skin, kidney, stomach, and small intestine. Transcripts were absent in testis, brain, heart, and skeletal muscle. The absence of a transcript in these latter tissues despite low activity being recorded is likely to be due to a low abundance of cells expressing TRAP mRNA relative to other tissues examined.
Examination of mouse tissues for TRAP protein revealed that, in bone, activity was associated with osteoclasts abutting in the vicinity of the mineralized surfaces (Figure 3A). In the skin, some activity was seen in the epidermis and in cells surrounding the hair follicles (Figure 3B). TRAP knockout controls showed no activity in either tissue. Further examination of the skin using immunohistochemistry identified TRAP protein in cells of the basal epidermis (Figure 3F) and in regions of the developing papillae (Figures 3F and 3G). Control skin stained with nonimmune serum did not show positive signals in these cells (Figure 3E). The pattern of TRAP protein in the skin resembled that expected for dendritic and Langerhans cells. Sections of skin were stained using an antibody to CD80 (B7) expressed on antigen-presenting cells. A strong positive reaction was produced in the same regions of skin that were positive for TRAP protein (Figures 3H and 3I).
Histochemical staining was weakly positive in peritubular regions of the kidney, but immunohistochemical staining gave equivocal results because of nonspecific reactions throughout the renal medulla. However, the enzyme was undetectable by either technique in glomeruli, including the mesangia (not shown).
In the liver, immunohistochemical staining gave a positive reaction in hepatocytes surrounding the venous system (Figures 3C and 3D). In situ hybridization using radiolabeled riboprobes to TRAP identified a pattern of expression in the liver (Figures 4B and 4C) comparable with that of the protein in Figures 3C and 3D. In situ hybridization on sections of bone from a femur in the metaphyseal region revealed expression of TRAP mRNA in chondrocytes within the growth plates and on the trabecular surfaces at the site of osteoclast activity (Figure 4A). In the spleen (Figures 4D and 4E), expression of mRNA was chiefly in sinusoidal cells surrounding the blood spaces. In the lung, TRAP expression was widespread (Figures 4F and 4G). Abundant expression was observed in alveolar cells including macrophages. In the skin (Figures 4H and 4I), expression of TRAP mRNA was concentrated in cells surrounding the dermal papillae and hair follicles as well as throughout the outer epithelial surface, consistent with the findings of the immunohistochemistry studies. Figure 4H is a control for skin hybridized with the TRAP sense riboprobe. Control sections for all other tissues hybridized with the sense riboprobe were negative and are not shown.
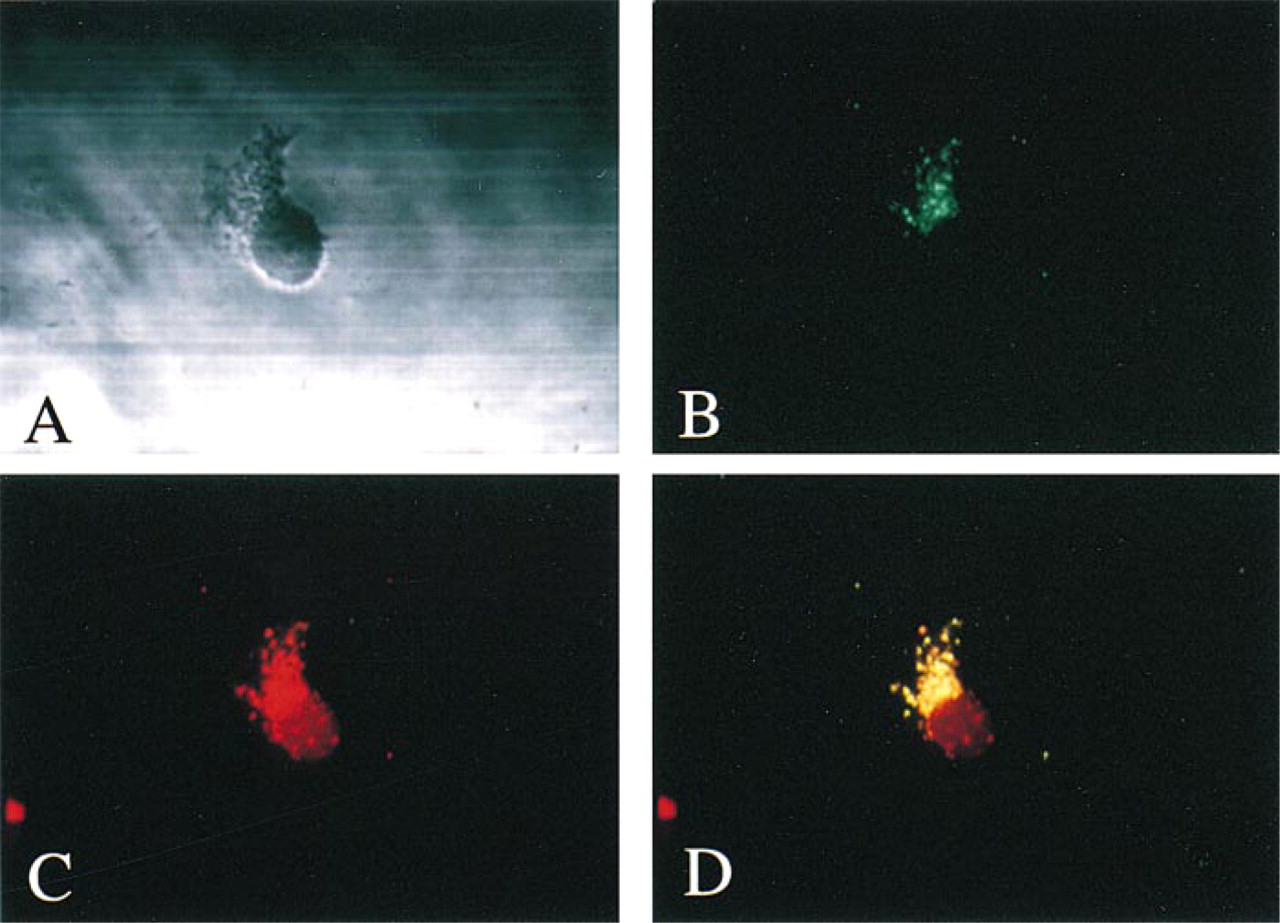
To investigate the expression of TRAP in bone marrow-derived antigen-presenting cells of dendritic phenotype, dendritic cells were freshly isolated from murine bone marrow by in vitro culture. As shown in Figure 5, confocal fluorescence microscopy of these cells showed positive staining with polyclonal antisera specific for the CD80 antibody, which recognizes the cell surface antigen representing the co-stimulatory B7 molecules that characterize antigen-presenting cells of dendritic lineage. These cells also demonstrated immunostaining with the isozyme-specific antibody directed against uteroferrin. Image superimposition of the two fluorescent antibody reactions confirmed that cells with plasma membrane staining for CD80 displayed punctate intracellular staining for TRAP.
Discussion
The iron-containing Type 5 acid phosphatase, TRAP, is a useful marker for osteoclasts and macrophages. Histochemically, osteoclasts show intense staining, whereas macrophages normally express TRAP at very low levels. However, increases in TRAP activity in specialized macrophages are characteristic of certain disease states, such as hairy cell leukemia and Gaucher's disease. Some previous studies have demonstrated that, in the osteoclast, TRAP is lysosomal (Reinholt et al. 1990), and other studies have shown that it is not in the lysosomes (Halleen et al. 1999). Here we show that in the macrophage it is lysosomal, as would be predicted from the presence of a putative N-terminal lysosomal leader sequence (Lord et al. 1990). We have demonstrated that, in the mouse, TRAP is widely distributed in tissues including bone, liver, spleen, lung, heart, kidney, stomach, small intestine, and skin. This wide distribution of TRAP expression corresponds to that predicted for phagocytic cells of the dendritic type. Here we show that TRAP expression is abundant in dendritic cells, as demonstrated by their characteristic distribution, their morphology, and specific cell surface immunostaining. Our studies show that TRAP activity attributable specifically to the Type 5 isozyme was present at sites where abundant Acp 5 mRNA expression was identified by in situ hybridization. Specific immunostaining for murine TRAP was also easily demonstrable in characteristic cells at these widely diverse tissue sites. The distribution immediately raises questions and sheds light on the general function of this highly conserved enzyme in specialized cell systems, including the Gaucher cell, and also in alveolar macrophages, which are highly phagocytic. The enzyme is secreted from osteoclasts during bone resorption and from macrophages. This, together with its lysosomal location in the macrophage, suggests that it has a digestive function.
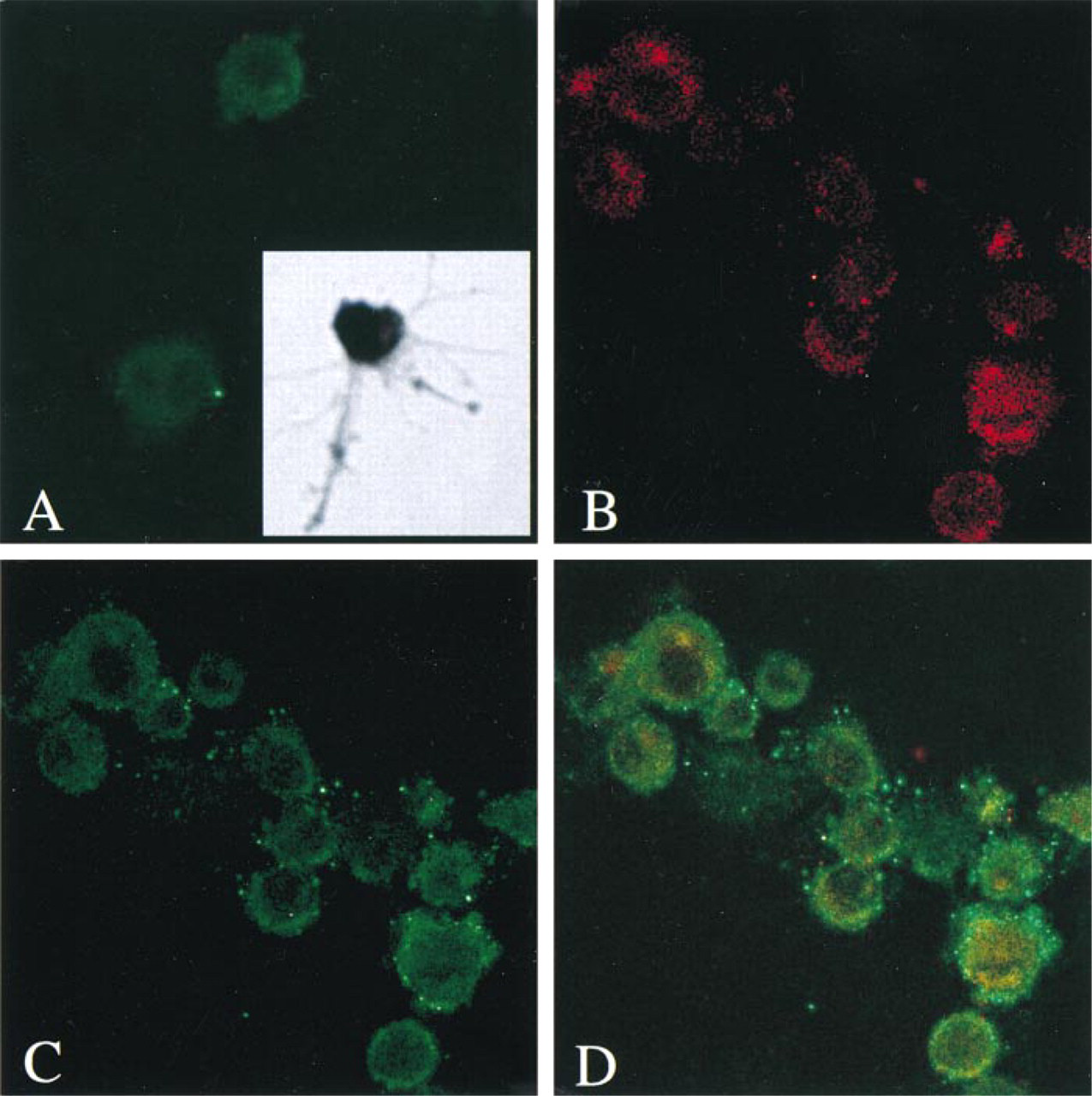
Identification of TRAP in bone marrow-derived murine dendritic cells. (
As demonstrated here, TRAP is also widely distributed in murine tissues. Abundant TRAP RNA transcripts were identified in skin and thymus as well as in relation to gastrointestinal epithelial surfaces, a distribution that corresponds closely to the known distribution of dendritic cells. Specific antibody reactions here subsequently confirmed co-expression of TRAP in murine dendritic cells and in cells of the predicted lineages. TRAP has long been associated with the hairy cells of leukemic endotheliosis (Bevilaqua et al. 1991) and serves as a routine histochemical marker for these immune cells, which exhibit somatic immunoglobulin gene rearrangements as well as phagocytic properties (Janckila et al. 1995). Hairy cell leukemia has been considered to represent a neoplastic proliferation of human dendritic cells (Jacobson et al. 1980; Jacobson personal communication 1990, 1999), and therefore our demonstration of TRAP expression in mouse dendritic cells may provide unifying cytochemical evidence for their common ontogeny.
The presence of enzymatically active TRAP protein in bone marrow-derived dendritic cells and macrophages that serve as antigen-presenting cells immediately raises questions as to its cytological function beyond simple degradation. Clearly, TRAP may retain its putative role in intralysosomal digestion in the processing of complex macromolecular antigens before presentation in the context of surface MHC Class II molecules. Because this process is dependent on the presence of cathepsin S specifically to cleave the invariant MHC Class II chains for surface display of antigenic peptides, and because activation of TRAP occurs by limited endopeptidase cleavage (Ljusberg et al. 1999), TRAP may also be activated in situ by the action of local proteinases of this class.
In the light of the observations reported, we are at present unclear as to whether the role of TRAP in dendritic cells is solely related to phosphoprotein degradation or whether it serves to regulate intracellular signaling pathways and specific functions including trafficking of MHC Class II-containing vesicles involved in antigen presentation.
Footnotes
Acknowledgments
Supported by the Medical Research Council and by a joint Biotechnology Award from the European Commission. ARH is in receipt of a Research Fellowship from the Arthritis Research Campaign.
We thank Dr Catherine Shanahan for advice with in situ hybridization and Dr Mike Allison for advice with culturing dendritic cells. Joan Grantham and Philip Ball kindly prepared the manuscript and figures.