
Abstract
Nervous system development is reliant on neuronal pathfinding, the process in which axons are guided to their target cells by specific extracellular cues. The ability of neurons to extend over long distances in response to environmental guidance signals is made possible by the growth cone, a highly motile structure found at the end of neuronal processes. Growth cones detect directional cues and respond with either attractive or repulsive movements. The motility of growth cones is dependent on rapid reorganization of the actin cytoskeleton, presumably mediated by actin-associated proteins under the control of incoming guidance signals. This article reviews how one such family of proteins, the ADF/cofilins, are emerging as key regulators of growth cone actin dynamics. These proteins are essential for rapid actin turnover in a variety of different cell types. ADF/cofilins are heavily co-localized with actin in growth cones and are necessary for neurite outgrowth. ADF/cofilin activities are regulated through reversible phosphorylation by LIM kinases and slingshot phosphatases. LIM kinases are downstream effectors of the Rho GTPases Rho, Rac, and Cdc42. Growing evidence suggests that extracellular guidance cues may locally alter actin dynamics by regulating the activity of LIM kinase and ADF/cofilin phosphatases via the Rho GTPases. In this way, ADF/cofilins and their upstream effectors may be pivotal to our understanding of how guidance information is translated into physical alterations of the growth cone actin cytoskeleton.
T
In recent years a myriad of guidance molecules have been identified (Goodman 1996; Tessier-Lavigne and Goodman 1996). Classification of these molecules by their activities has proved more difficult. It appears that a number of different factors come into play in the decision of whether a guidance molecule will behave as an attractive or a repulsive cue. For example, the level of cAMP in the growth cone can determine if the same cue attracts or repels it (Song and Poo 1999). Results from a number of studies have firmly established that growth cone motility is mediated by the actin and microtubule cytoskeletons. A number of excellent reviews have summarized the role of cytoskeletal dynamics in neuronal pathfinding (Bray and Hollenbeck 1988; Mitchison and Kirschner 1988; Bentley and O'Connor 1994; Lin et al. 1994; Tanaka and Sabry 1995; Heidemann 1996; Letourneau 1996). Most guidance molecules exert their effects through rapid reorganization of the actin cytoskeleton. However, the actin binding proteins that elicit these changes and the signaling pathways that control their activity still demand further characterization. The first clues as to how actin-based cell motility is regulated came from Hall and colleagues (Ridley and Hall 1992; Ridley et al. 1992). These pioneering studies on fibroblasts demonstrated that a small family of GTP binding proteins, the Rho GTPases, were responsible for coordinating the formation of lamellipodia and filopodia. Both of these actin-rich structures constitute the motility machinery of migrating cells and are found on the periphery of growth cones.
The highly conserved family of actin-associated proteins, the actin depolymerizing factors (ADFs) and cofilins, are involved in regulating actin dynamics in the growth cone. Although ADFs and cofilin are products of different genes and differ quantitatively in their interactions with G- and F-actin (Vartiainen et al. 2002; Yeoh et al. 2002), the proteins have very similar qualitative effects on actin dynamics, are regulated on the same site by reversible phosphorylation, and are usually co-localized in cells. Therefore, we will consider them here as a single entity (ADF/cofilin) that we will refer to as AC. AC is essential for the rapid turnover of actin filaments in vivo and stimulates cell growth and motility in a variety of cell types (Bamburg 1999). In neurons, AC is expressed at high levels and co-localizes with F-actin in the growth cone (Bamburg and Bray 1987). Furthermore, overexpression of AC in neurons leads to increased neurite outgrowth (Meberg and Bamburg 2000). LIM kinases, important regulators of AC, are downstream targets of the Rho GTPases, suggesting that AC may be a target of extracellular signals that regulate growth cone behavior (Kuhn et al. 2000). This review discusses how AC may be one key to our understanding of how the influences of guidance molecules are translated via signaling pathways to physical changes in the growth cone actin cytoskeleton.
Actin Cytoskeletal Dynamics in the Growth Cone
The diverse functional roles of the actin cytoskeleton in the many cellular processes in which it is involved are made possible by the ability of actin filaments to form a variety of distinct assemblies with specific biophysical and biochemical properties. The formation of such assemblies is brought about by the interactions of actin monomers (G-actin) and filaments (F-actin) with a catalogue of actin binding proteins that serve to anchor, crosslink, or regulate the polymerization status of the actin network in the cell. Under physiological conditions, monomeric units reversibly polymerize into non-covalent filaments as ATP-bound actin (Figure 1). Shortly after incorporation of ATP-bound subunits into the filament, ATP is hydrolyzed to ADP-Pi-actin. The loss of inorganic phosphate is coupled to a change in the filament actin conformation (Belmont et al. 1999). At steady state, this ADP-actin constitutes the pointed end of F-actin, and the ATP-actin makes up the barbed end of the filaments. The concentration of monomer needed to maintain growth at the barbed end of the filament is less than that needed to maintain growth at the pointed end, and subunits are therefore lost from the pointed end as the barbed end grows. This process, known as treadmilling, results in a net flux of subunits through the filament. During treadmilling at steady state, any single filament can grow or shrink but the polymer mass remains constant.
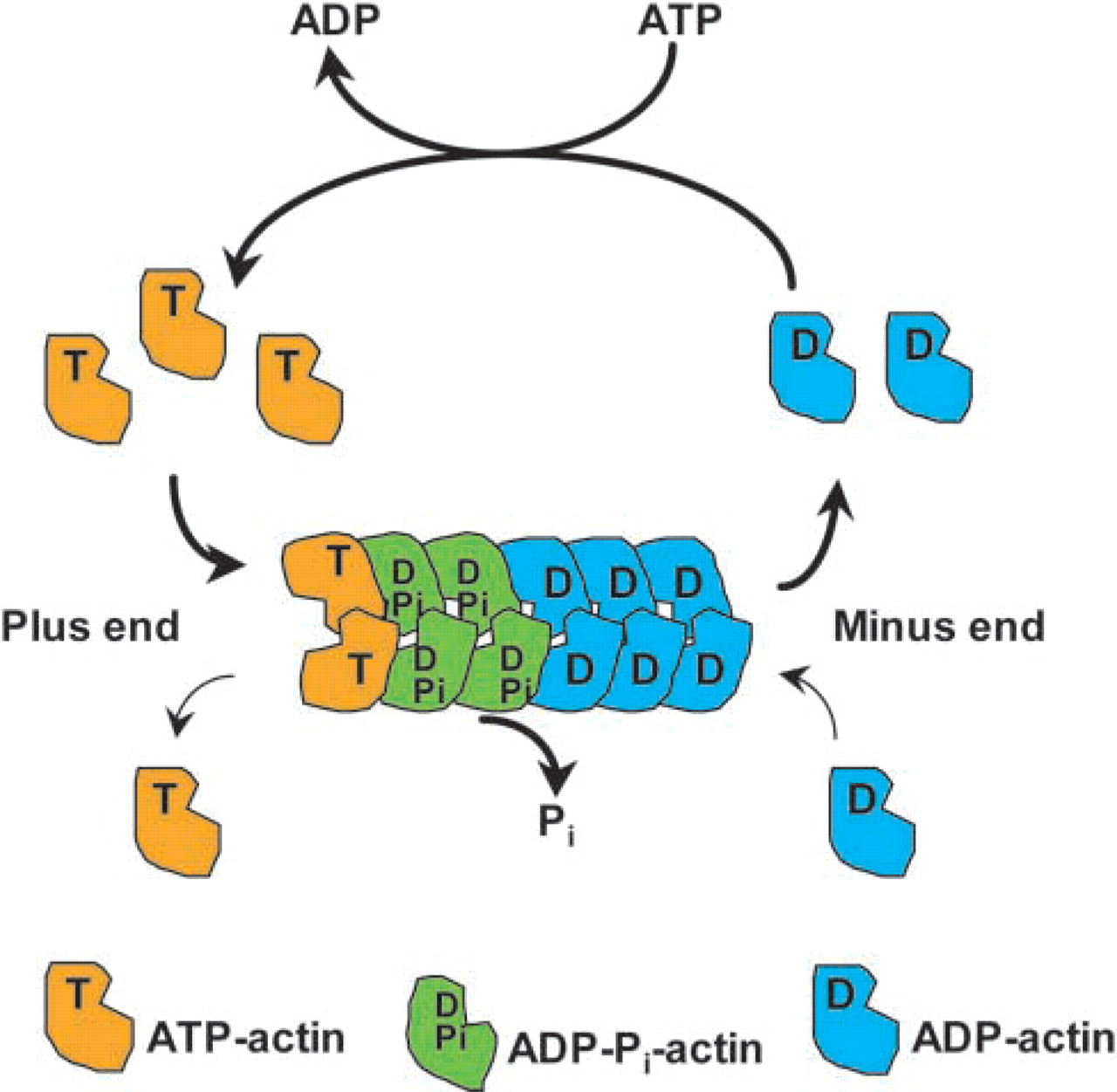
Treadmilling cycle of actin filaments at steady state. At steady state, subunits undergo net assembly at the plus end of the filament and net disassembly at the minus end at an identical rate. Therefore, even though there is a net flux of subunits through the filament, the net rate of filament growth is zero. This process is known as treadmilling.
To understand how the interaction of growth cones with a guidance molecule can alter turning, it is first necessary to understand growth cone structure and cytoskeletal organization (Figure 2). The peripheral domain of the growth cone consists of a layer of flat web-shaped sheets of cytoplasm called lamellipodia, which surround the central domain of the growth cone. Finger-like protrusions called filopodia extend from the outer edge of the peripheral domain, connected to each other by lamellipodia (Forscher et al. 1987). Many actin bundles protrude longitudinally into filopodia and extend back into lamellipodia, where they form a mesh-work of actin filaments (Bridgman and Dailey 1989; Letourneau 1983). The actin bundles that ramify filopodia are arranged in a unipolar fashion, with the barbed ends of at least most filaments facing outwards towards the cell membrane. Lamellipodia of the more highly organized growth cones of Helisoma and Aplysia contain an actin meshwork of more uniform polarity (Welnhofer et al. 1997). Conversely, actin filaments in lamellipodia observed in dorsal root ganglion and other mammalian growth cones are arranged into an orthogonal array with less distinct polarity (Lewis and Bridgman 1992). Both lamellipodia and filopodia are central to growth cone motility and undergo continuous cycles of expansion and contraction (Bray and Chapman 1985; Goldberg and Burmeister 1986). The turnover of actin filaments in these structures is very high. Exposure of growth cones to cytochalasin B, a drug that caps the barbed ends of filaments and thus prevents monomer addition (MacLean-Fletcher and Pollard 1980; Bonder and Mooseker 1986), results in rapid breakdown of the growth cone actin cytoskeleton (Forscher and Smith 1988).
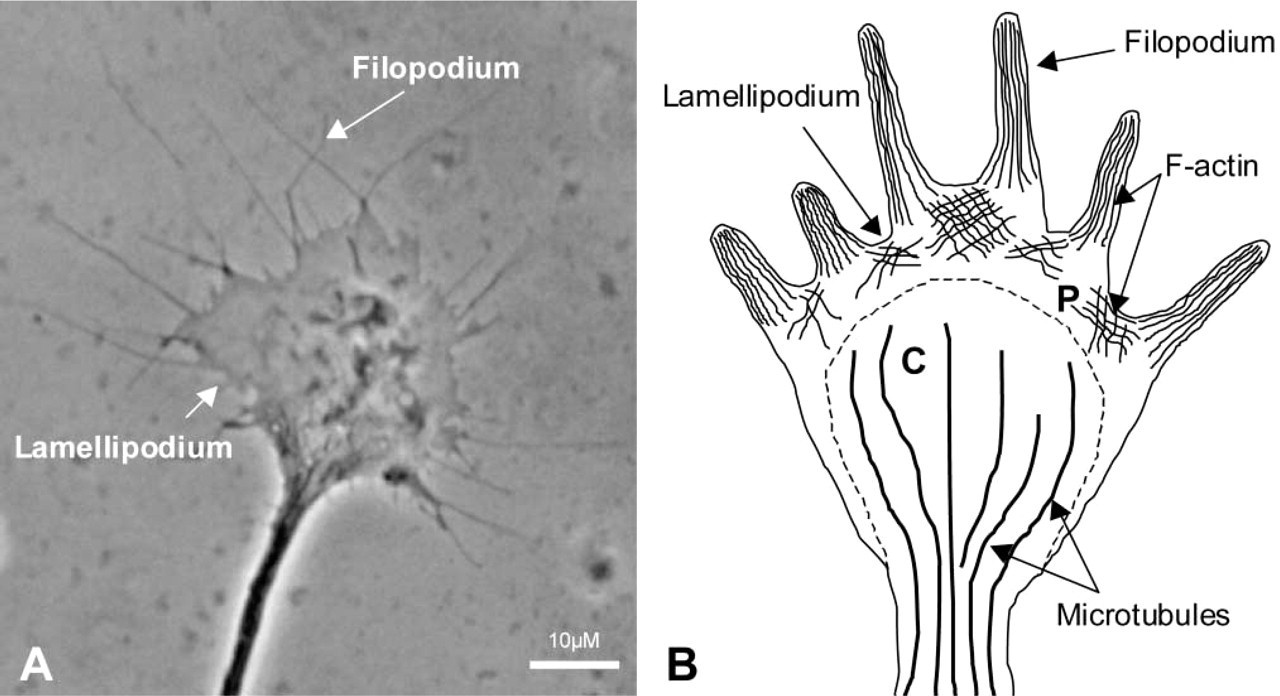
The neuronal growth cone. (
The mechanisms by which lamellipodia and filopodia extend and retract involve a number of actin binding proteins that are localized and regulated in specific regions of the leading edge of migrating cells. Examination of lamellipodia in keratocytes and fibroblasts by electron microscopy (Svitkina and Borisy 1999) revealed an extensively branched array of actin filaments, known as the dendritic brush, at the leading edge. A treadmilling model for the entire actin array was proposed (Svitkina and Borisy 1999) (Figure 3). Although the actin network at the leading edge of growth cones has not been so well characterized, it is very likely that many elements of the fibroblast model also exist in neurons. The motility of growth cones is dependent on the assembly, disassembly, and retrograde flow of an organized array of filamentous actin at the leading edge (Lin and Forscher 1995). These three kinetically independent processes rely on the coupling of the actin cytoskeleton to external substrates. This coupling may be achieved by membrane cytoskeletal “clutch” proteins that link the actin network to the extracellular substrates. Therefore, the rate of neurite extension may be governed by the extent of clutch protein-mediated actin-substrate linkage.
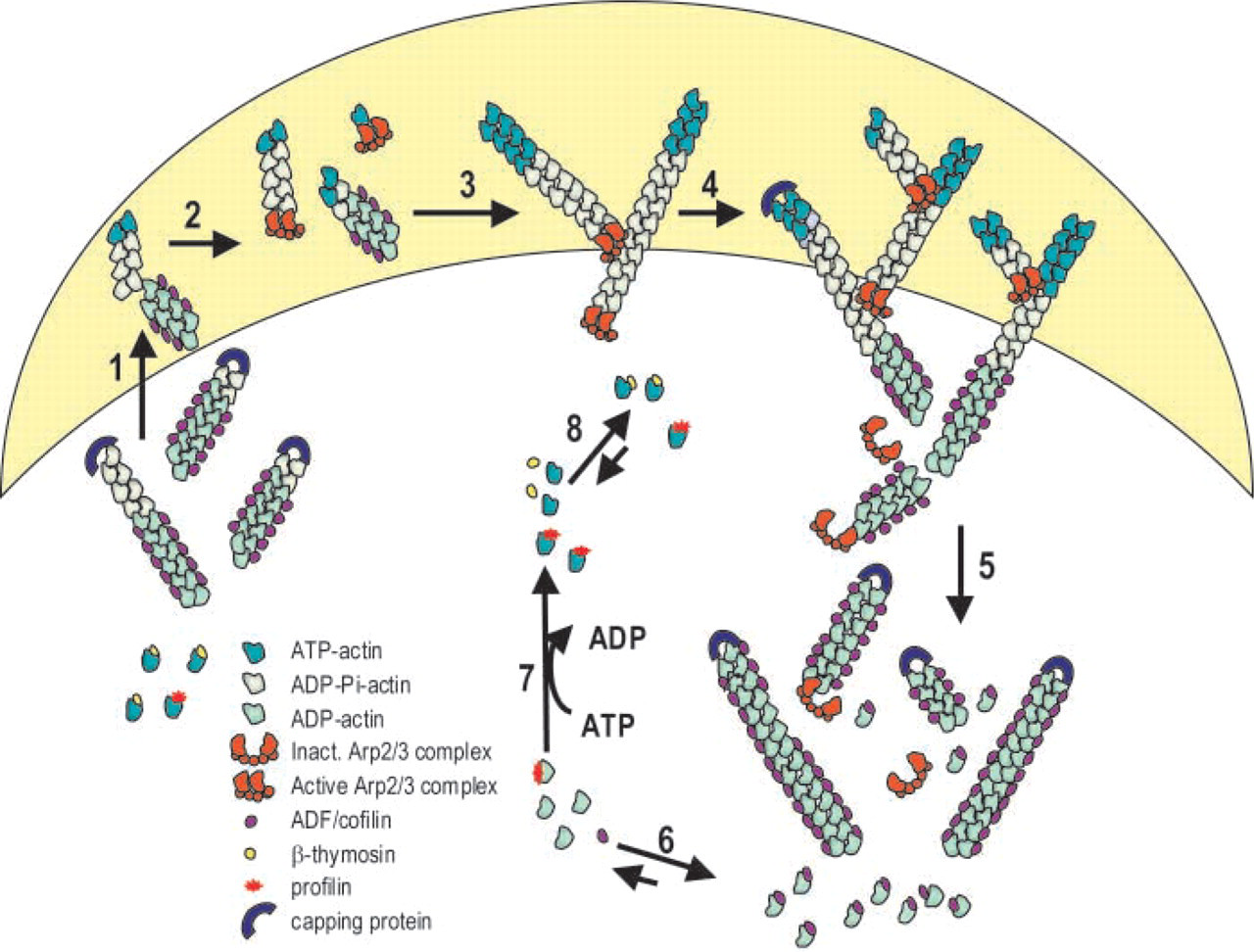
Model for the turnover of the actin filament array at the leading edge of lamellipodia. The cytoskeletal network at the leading edge of motile cells consists of an extensively branched array of actin filaments with barbed ends facing forward and pointed ends making Y-junctions (about 70° angles) with other filaments. These Y-junctions are formed by the Arp2/3 complex (Svitkina and Borisy 1999). The treadmilling of the dendritic array is regulated by a number of actin binding proteins as described below. Numbers in parentheses refer to numbered steps in the diagram. (1) The barbed ends of actin filaments are usually capped by capping protein when the cell is in resting state. When cells are stimulated, free barbed ends are generated in the leading edge (shaded area) to nucleate filament growth and drive membrane protrusion and cell movement. Three potential mechanisms exist for the generation of free barbed ends (Condeelis 2001): dissociation of capping proteins by elevated levels of Ptdlns(4,5)P2, filament severing by ADF/cofilin, and actin nucleation dependent Arp2/3. The Arp2/3 complex is activated on binding to WASP, one form of which is activated by Cdc42. (2) The active Arp2/3 complex nucleates actin filament assembly and/or caps the free pointed end of the ATP- or ADP-Pi-actin filaments (Machesky et al. 1999; Blanchoin et al. 2000; Pollard et al. 2001). (3) The activated Arp2/3 complex binds to the side of the filament and then nucleates filament growth or captures pointed ends of a preexisting filament (Bailly et al. 1999; Blanchoin et al. 2000). An alternative hypothesis proposes that the activated Arp2/3 complex binds to and branches the barbed end of actin filaments (Pantaloni et al. 2000). Growth of filaments is rapid and the lag in Pi disassociation leads to filaments in the leading edge that are composed mostly of ATP and ADP-Pi actin and do not bind ADF/cofilin. (4) At the rear of lamellipodia, two mechanisms may contribute to rapid depolymerization: filament severing or uncapping of pointed ends by removal of the Arp2/3 complex. Severing by AC likely occurs at junctions between regions of filaments that are saturated with AC and naked F-actin. The activated Arp2/3 complex disassociates from the pointed end of ADP-actin because of its weaker affinity for ADP-actin than for ATP- or ADP-Pi-actin (Blanchoin et al. 2000). (5) Capping of barbed ends by capping proteins prevents their further elongation. AC enhances depolymerization of ADP-actin from free filament ends in the rear of the lamellipodia. (6) The complex of AC and ADP-actin that disassociates from free filaments ends is in equilibrium with AC and ADP-actin monomer. (7) The nucleotide exchange on actin monomer is a slow process, further inhibited by AC. Profilin enhances nucleotide exchange (Nishida 1985; Blanchoin and Pollard 1998; Didry et al. 1998). (8) ATP-actin monomers are sequestered by β-thymosin (Sun et al. 1995) to prevent spontaneous nucleation, but provide a pool of ATP-actin for assembly.
The assembly of actin filaments is believed to result from a combination of nucleation, polymerization, and annealing of short filaments, although which of these processes predominates in growth cones remains unclear. Actin elongation in this region generates the force necessary to drive lamellipodial and filopodial extension. Filament assembly may be brought about by nucleation factors such as the Arp2/3 complex (Welch et al. 1997a,b), uncapping of barbed ends by capping proteins (Condeelis 2001), and the activity of actin monomer-sequestering proteins such as profilin, thymosin β-4, and twinfilin (Goldschmidt-Clermont et al. 1991; Pantaloni and Carlier 1993; Palmgren et al. 2002). Profilin can enhance actin assembly by catalyzing the conversion of ADP-actin to ATP-actin, a form that assembles more rapidly and has slower dissociation kinetics. Proteins of the Ena/Mena/VASP family may also increase polymerization by recruiting profilin-actin to sites of actin assembly (Chakraborty et al. 1995; Gertler et al. 1996; Kang et al. 1997). Ena/Mena/VASP proteins play a key role in cortical neuron positioning during development through their spatial control of actin assembly. Mena is essential for neural development in mice (Goh et al. 2002) and is concentrated at the tips of growth cone filopodia, the site of actin assembly (Bear et al. 2002).
AC proteins may also contribute to nucleation by severing growing actin filaments, thereby generating new filament ends. Whether AC proteins promote filament assembly or disassembly depends on a number of different factors. For example, if the filaments were capped or tethered at the pointed ends, dissociation of monomers would be prevented, allowing net filament assembly to occur from the barbed ends.
After assembly at the leading edge, bundles of actin move back to the central domain of the growth cone via myosin-mediated retrograde F-actin flow (Forscher and Smith 1988; Okabe and Hirokawa 1991; Lin et al. 1996; and Brown and Bridgman, this issue). Actin filaments are then disassembled at the rear of the peripheral domain, allowing monomers to be recycled for further rounds of polymerization (Forscher and Smith 1988). The destruction of actin filaments may be mediated by filament-severing proteins such as gelsolin (Yin and Stossel 1980) and/or depolymerizing factors such as AC (Bamburg 1999). The dynamic nature of growth cones enables them to respond to guidance cues in the extracellular environment and to react with either repulsive or attractive movements. Such actions are dependent on the asymmetric collapse of the growth cone in a specific area. This process, sometimes known as “growth cone turning,” is associated with a loss of actin organization in the region of collapse and the assembly of filaments in the direction of movement.
Regulation of Actin Dynamics by ADF/Cofilin in the Growth Cone
Several lines of evidence point towards AC as a crucial protein in growth cone motility. This section focuses on how the unique activities of AC may serve to mediate the rapid changes in actin dynamics observed in growth cones. The depolymerizing activity of ACs arises from their ability to increase the rate of dissociation of ADP-actin from the pointed end of actin filaments (Carlier et al. 1997) and to increase the number of filament ends through fragmentation or severing (Maciver 1998). Actin filaments turn over at a rate 100–200-fold faster in vivo than in vitro, the difference requiring AC proteins (Zigmond 1993).
Animal ACs are regulated by phosphorylation on a highly conserved serine residue (Agnew et al. 1995). The phosphorylated form of AC is inactive in actin binding and depolymerizing assays (Morgan et al. 1993) but can be reactivated by dephosphorylation. The activation of AC from the phosphorylated pool occurs in response to stimulation in many cell types, suggesting that changes in the activity of AC might be regulating actin dynamics in many of the cellular phenomena examined (Bamburg 1999). However, net changes in the phosphorylation/dephosphorylation of AC to enhance actin turnover from the pointed ends of filaments may not be required. Because AC binding to ADP-actin inhibits nucleotide exchange, phosphorylation of AC that dissociates from the complex will prevent its re-association and accelerate nucleotide exchange on actin, thus promoting actin's ability to reassemble (Bernstein et al. 2000). A coordinated dephosphorylation of the phosphorylated AC (pAC) will reactivate it to remove another subunit and drive filament turnover (Figure 4, steps 1–5). Therfore, a net change in phosphorylation of AC may not be as critical to actin dynamics as is the turnover rate of the pAC pools (i.e., phosphocycling). Evidence for AC phosphocycling in non-neuronal cells has been presented (Meberg et al. 1998). In this way, ACs possess the properties to enhance actin turnover at the leading edge of migrating cells, including growth cones.
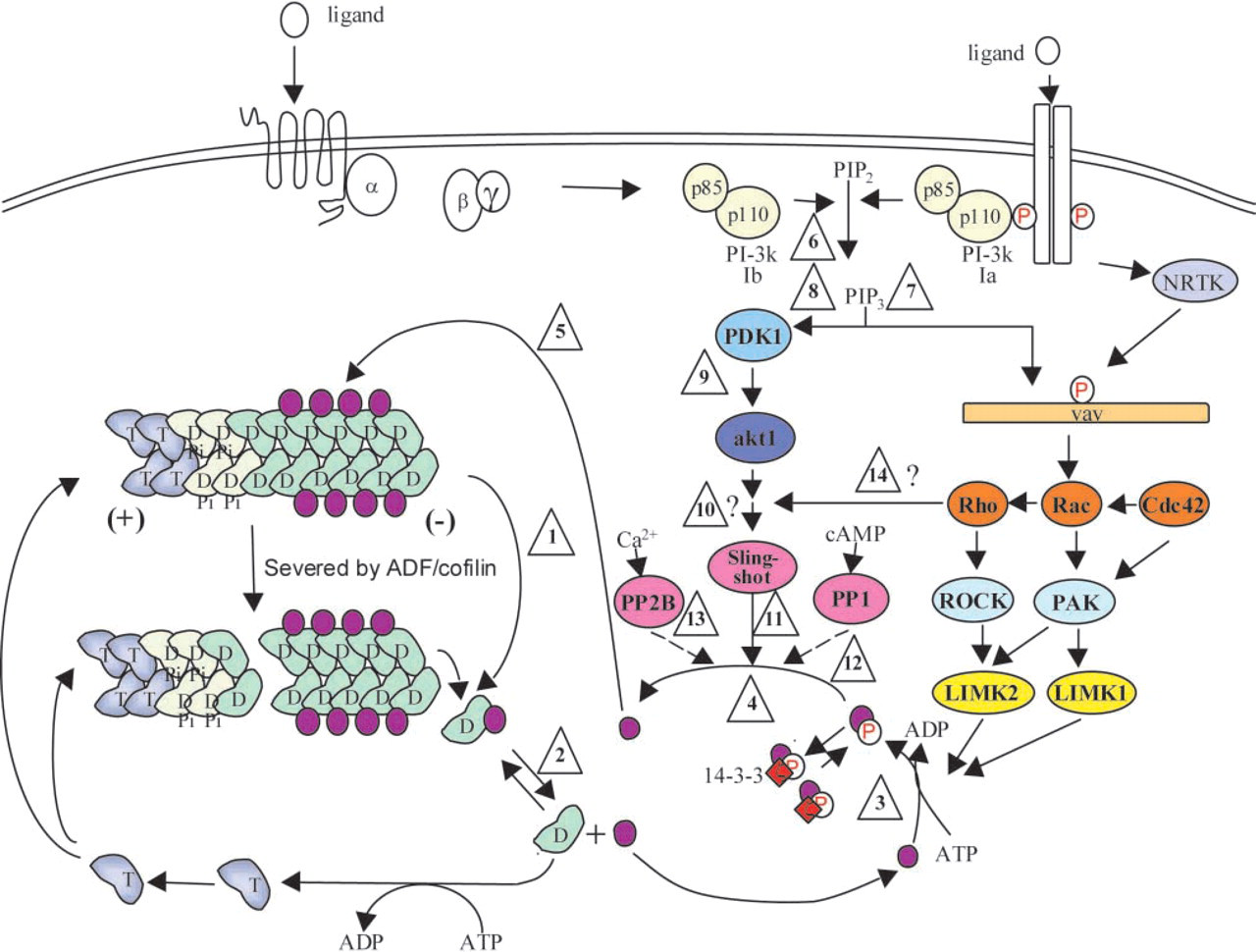
Speculative model for the regulation of actin dynamics through AC phosphocycling. The phosphocycling pathway is shown in steps 1–5. (1) AC enhances the depolymerization of actin filaments (T, ATP-actin; D, ADP-actin) both by severing and generating more filament ends and by enhancing the off rate at the pointed (-) end of actin. (2) The AC-ADP-actin complex dissociates with a Kd ∼150 nM, leaving the free ADP-actin to exchange nucleotide. (3) AC is phosphorylated by LIM kinases, considerably reducing its affinity for either G- or F-actin. (4) Phosphorylated AC is reactivated by one of several different phosphatases. The degree of AC dephosphorylation may be affected by levels of 14-3-3ζ protein, which binds to the phosphoserine of AC, protecting it from phosphatase-mediated dephosphorylation. (5) Active AC binds to actin filaments, enhancing another round of depolymerization. The regulatory pathway is shown in steps 6–14. (6) A variety of extracellular ligands activate converging upstream pathways that activate PI3 kinase, producing PIP3 in the plasma membrane. This is proposed to be the first step in the bifurcating pathway for phosphoregulation of AC. (7) The PIP3 recruits adapter molecules (represented here by vav) that either possesses or recruits a guanine nucleotide exchange factor (GEF) for one of the Rho family of GTPases. Some adapter molecules may require further activation by non-receptor tyrosine kinases (NRTK) to expose the GEF domain. Actin Rho family GTPases stimulate specific kinases (PAK or ROCK) to phosphorylate one of the two isoforms of LIM kinase. (8) PIP3 also activates the PIP3-dependent kinase (PDK1), which (9) activates the protein kinase, akt (also called protein kinase B). We speculate that downstream of akt is a specific phosphatase, possibly of the slingshot family, that can activate ADF and cofilin through dephosphorylation (11). The less specific phosphatases PP1/PP2A (12) and PP2B (13) are activated by cAMP and calcium, respectively, to dephosphorylate ADF. Inhibition of PP1/PP2A with calyculin A results in a large net dephosphorylation of ADF and cofilin, suggesting that the active PP1 and/or PP2A dephosphorylate one or more of the intermediates in the PDK1/akt pathway. (14) The Rho GTPases may also be involved in regulating the pAC phosphatase, thereby enabling them to control the activation and inactivation of AC.
In addition to biochemical studies, in vivo observations have confirmed the importance of AC in neuronal development. ADF is expressed at high levels in neurons and co-localizes with F-actin in growth cones (Bamburg and Bray 1987). Adenovirus-mediated overexpression of Xenopus ADF (XAC) in rat cortical neurons caused a significant increase in neurite length (Meberg and Bamburg 2000). Moreover, enhanced XAC expression led to larger growth cones and more filopodia. The rate of actin depolymerization after treatment with latrunculin, an actin monomer-sequestering compound, was greater in growth cones overexpressing XAC than in control cells, suggesting enhanced filament turnover in these cells (Meberg and Bamburg 2000). It is very likely that an increase in actin treadmilling and turnover promotes lamellipodial and filopodial protrusion, thereby increasing the rate of growth cone advance and neurite extension. Accordingly, overexpression of a non-phosphorylatable but active analogue of ADF had very little effect on neurite length, whereas inactive XAC had none.
Rho GTPase Regulation of Neuronal Morphology
The Rho family of small GTPases (Rho, Rac, and Cdc42) activate various pathways that affect cell locomotion through dynamic regulation of the actin cytoskeleton. The activities of the Rho GTPases were first characterized by microinjection studies in fibroblasts. For example, Rho A stimulates the formation of stress fibers (Ridley and Hall 1992), Rac reorganizes actin into lamellipodia (Ridley et al. 1992) and Cdc42 induces the formation of filopodia (Nobes and Hall 1995). Rho GTPases bring about such changes by regulating adaptor proteins and kinases which, in turn, alter the activity of specific actin-modulating proteins. Expression of Rho GTPases is widespread in a range of neural tissues (Yamamoto et al. 1988; Olenik et al. 1999). The evident importance of these proteins in actin-based motility led investigators to characterize their functioning in neurons. Initial studies showed that Rac1 is essential for lamellipodial formation (Luo et al. 1994), axon outgrowth, and dendritic spine formation (Luo et al. 1996). Expression of dominant negative forms of Rac and Cdc42 during Drosophila neural development inhibits neurite formation, whereas expression of their constitutively active forms promotes neurite growth (Threadgill et al. 1997).
More recent findings also support the involvement of Rac and Cdc42 in growth cone protrusion. For example, constitutively active Rac and Cdc42 increase dendritic branching in Xenopus (Lee et al. 2000). Furthermore, laminin-1, a basement membrane glycoprotein, stimulates neurite outgrowth through activation of Cdc42 (Brown et al. 2000; Weston et al. 2000). Conversely, Rho-induced stress fiber formation leads to growth cone collapse and neurite retraction, presumably by sequestering actin for incorporation into stress fibers that would otherwise be available for lamellipodia and filopodia formation (Sebok et al. 1999; Lee et al. 2000; Li et al. 2000).
Other studies further investigated the role of small GTPases in growth cone collapse (Kuhn et al. 1999). Active forms of RhoA, Rac1, and Cdc42 were expressed in chick motor neurons. The cells were then exposed to either CNS myelin or collapsin-1, two factors that induce rapid growth cone collapse. Cells expressing constitutively active Rac1 and RhoA, but not Cdc42, were able to resume normal growth after treatment with CNS myelin and displayed no signs of collapse. In the case of collapsin-1, only cells expressing constitutively active Rac1 and Cdc42 blocked collapse. These results suggest that CNS myelin and collapsin-1 signal through different Rho GTPases to bring about growth cone collapse.
Rho GTPase Signaling to ADF/Cofilin in the Growth Cone
It is clear that the effects of Rho GTPases on growth cone morphology and motility require their signaling to specific actin-modulating proteins. At present, only one group of such proteins, the ADF/cofilins, appears to possess the necessary upstream effectors to account for the Rho GTPase-triggered actin reorganization observed in neuronal and non-neuronal cells. The kinases regulating AC in vertebrates are LIM kinase (LIMK) (Arber et al. 1998) and testicular protein kinase (TESK) (Toshima et al. 2001). Two isoforms of LIMK have been identified. LIMK1 is expressed predominantly in both the peripheral and central nervous systems (Arber et al. 1998), whereas LIMK2 is more ubiquitous (Sumi et al. 1999). A developmentally regulated inactive species of LIMK1, missing 21 amino acids in the kinase domain but that can form dimers with LIMK1 and downregulate its activity, also exists (Bernard et al. 1994). Hemizygosity of the LIMK1 gene, which leads to abnormally low expression levels of LIMK1, is associated with the visual-spatial cognitive disorder Williams syndrome (Donnai and Karmiloff-Smith 2000). LIMK1 null mutants of mice develop learning impairments (Meng et al. 2002), further emphasizing the importance of LIMK1 in correct neural development.
Both LIMK1 and LIMK2 are downstream effectors of the Rho GTPases (Figure 4, steps 6–14). LIMK1 and LIMK2 are regulated by Rac, Cdc42, and Rho (Arber et al. 1998; Yang et al. 1998; Maekawa et al. 1999; Sumi et al. 1999). Rac and Rho are activators of the p21-activated kinase (PAK) and Rho-associated kinase (ROCK), respectively (Manser et al. 1994; Matsui et al. 1996). The former activates LIMK1 and the latter activates LIMK2 (Edwards et al. 1999; Maekawa et al. 1999). Dominant negative LIMK1 blocks Rac-stimulated lamellipodial protrusion (Yang et al. 1998), Cdc42-induced filopodia formation, and Rho-mediated stress fibers (Sumi et al. 1999). Because ADF and cofilin are the only known physiological substrates of the LIMKs, it is reasonable to assume that all members of the Rho family of GTPases alter actin dynamics, at least in part, through the regulation of AC proteins.
The few studies that have sought to address the link between Rho GTPases and AC proteins in neurons have yielded some interesting results. LIMKs contain two amino-terminal LIM domains, a central PDZ domain, and a carboxyl-terminal kinase domain with serine/threonine kinase activity. Overexpression of the noncatalytic N-terminal domains of LIMK1 decreased neurite extension after stimulation with NGF and Rhokinase inhibitor but had no effect on spontaneous neurite outgrowth (Birkenfeld et al. 2001). These results suggest that the LIM domains either play an important role in regulating the kinase activity of the enzyme by somehow interacting with elements of signaling pathways, in this case downstream effectors of NGF and Rho-kinase inhibitor, or have some other scaffolding effects in signaling pathways.
Other studies have shown that the phosphorylation state of AC in neurons is influenced by growth factors as well as elements of second messenger systems. Elevations in intracellular calcium and cAMP levels bring about extensive dephosphorylation of ADF in cortical neurons (Meberg et al. 1998). Growth factors have dual effects on AC activity in neuronal and non-neuronal cells. The phosphorylation state of AC during NGF-induced neurite extension was monitored in PC-12 cells (Meberg et al. 1998). Before NGF addition, ADF and cofilin were mostly phosphorylated and spread diffusely throughout the cytoplasm. At 10 min after NGF addition, a dramatic dephosphorylation of the ADF and cofilin pools occurred and both proteins colocalized with actin at the tips of lamellipodia and filopodia. The elevated levels of dephosphorylated ADF and cofilin persisted for a further hour, as did the co-localization of ADF and cofilin with actin in membrane ruffles. These results suggest that active AC is needed to mediate actin dynamics in motile regions of the cell.
It is important to note that net AC dephosphorylation is not always necessary for growth factor-induced lamellipodial and filopodial extension. When fibroblasts were treated with EGF, rapid lamellipodial growth was observed (Meberg et al. 1998). Despite these increases in actin dynamics there was no net change in the level of phosphorylated AC, but the rate of turnover of phosphate on AC was increased considerably. These results indicate that a balance between kinase and phosphatase pathways exists to maintain the cycling of phosphate on AC. Dephosphorylation of AC is regulated in part by the exposure of the phosphoserine, which can be bound to a specific isoform (ζ) of 14-3-3 protein shielding its dephosphorylation from general phosphatases (Gohla and Bokoch 2002). Furthermore, it is unlikely that the more general phosphatases, such as PP1 or PP2A/2B, could be so precisely coordinated with LIMK activity. In agreement with this theory, evidence for an AC phosphatase that is not inhibited by inhibitors of PP1 or PP2A/2B has been confirmed by several studies (reviewed in Bamburg 1999). The treatment of intact cells with calyculin or okadaic acid, inhibitors of PP1 and PP2A, results in an almost complete dephosphorylation of pAC. This implies that these phosphatases (PP1/PP2A) are involved in the dephosphorylation of some protein that, when phosphorylated, enhances the dephosphorylation of pAC.
Although the AC phosphorylation pathway occurs through Rho activation of LIMK1, the regulation of the dephosphorylation is less well understood. Recently, an ADF/cofilin phosphatase called slingshot was identified (Niwa et al. 2002). However, its regulation is unknown. Expression of constitutively active Akt (also known as protein kinase B) increases AC dephosphorylation in 3T3 L1 cells (Okada et al. 1999), suggesting that the AC phosphatase may lie downstream of this signaling molecule. Alternatively, it is quite possible that the AC phosphatase is regulated by kinases downstream of the Rho GTPases. Rapid phosphate turnover on AC would allow its activity to be tightly regulated in specific regions of the growth cone actin cytoskeleton. Growth cone turning is dependent on the spatially coordinated creation and destruction of actin-based structures. This may result from local activation and inactivation of AC in the growth cone by phosphatase and kinase to facilitate actin assembly in one region and its disassembly in another.
Only one study has shown how signaling from an extracellular guidance cue can directly influence the behavior of AC proteins in neurons. Aizawa and colleagues (2001) demonstrated that the extracellular guidance cue semaphorin 3A (sema 3A) signals downstream to cofilin via LIMK1 to promote reorganization of the growth cone actin cytoskeleton. Sema 3A, originally known as collapsin-1, behaves as either a repulsive or an attractive guidance cue and is capable of causing collapse in dorsal root ganglion growth cones. After sema 3A treatment, cofilin phosphorylation was observed to first rapidly increase, then gradually decrease at the collapsing growth cone. The changes in cofilin phosphorylation state seemed to correspond with an initial assembly and subsequent disassembly of actin filaments in the growth cone. The transient phosphorylation and subsequent dephosphorylation of cofilin after sema 3A treatment suggests the existence of a cofilin phosphatase. Although inhibition of LIMK1 activity blocked sema 3A-induced growth cone collapse, other experiments demonstrated that this effect was not exclusively due to LIMK. Expression of constitutively active and dominant negative LIMK1 had no effect on growth cone morphology (Aizawa et al. 2001). This finding strongly suggests only the combined activities of cofilin kinase and phosphatase can bring about collapse after sema 3A treatment. The reactivation of cofilin by a phosphatase would promote filament turnover thereby inducing growth cone collapse.
Conclusion
The evidence presented in this review clearly defines AC proteins as essential regulators of neuronal growth cone actin dynamics. The current models for actin-based cell motility all require fast turnover of actin filaments at the leading edge. The unique ability of AC proteins to increase the rate of monomer dissociation from filaments may, in conjunction with other actin-associated proteins, drive the rapid actin filament dynamics observed in neuronal growth cones and other migrating cells. Actin reorganization in growth cones appears to be controlled by the Rho GTPases, which function downstream of at least some attractive or repulsive extracellular guidance molecules. There is now a definitive link between signaling from one guidance cue to AC proteins. AC regulation by phosphorylation appears to be a crucial way in which these proteins are regulated. AC functioning in growth cones is dependent on a balancing of kinase and phosphatase pathways. The recent identification of the slingshot family of AC phosphatases warrants the identification and localization of their neuronal isoforms. This would allow the effect of partial loss- and gain-of-function mutants of AC phosphatase on the growth cone actin cytoskeleton to be assessed, as well as to determine the mechanism of slingshot regulation. Subsequent studies could then focus on how the combined activities of AC phosphatase and LIM kinase mediate the changes in growth cone motility stimulated by guidance molecules. Turning responses result from the asymmetric collapse of the growth cone, a change associated with the assembly and disassembly of actin-based structures. It seems likely that these changes will involve the spatially coordinated regulation of AC proteins.
Footnotes
Acknowledgements
Supported in part by grants from the Alzheimer's Association (IIRG-01-2730) and the National Institutes of Health (GM35126, NS40371) to JRB.