
Abstract
We developed a simple and reliable technique for immunofluorescence detection of F-actin on microtome sections of plant tissues. For the first time, large numbers of plant cells from various tissues that pass through their developmental stages could be consistently visualized on one section from plant organs. n-Maleimidobenzoic acid N-hydroxy-succinimide ester-pretreated and formalin-fixed segments of plant roots and shoots were embedded in low melting point ester wax at 37C and sectioned on a microtome. After dewaxing and rehydration, microfilaments were visualized by indirect immunofluorescence technique with a monoclonal anti-actin antibody. The technique has been successfully used for visualization of tissue- and development-specific F-actin arrays in cells of Zea mays and Lepidium sativum root tips and of maize stem nodes.
S
Immunocytochemical techniques permit the visualization of specific isoform of actin. This is of great interest to plant biologists because of the high divergence in multigene families that encode plant actins (Baird and Meagher 1987). McLean et al. (1990) demonstrated tissue specific localization of Λ-actin and K-actin in soybean roots. In addition to the detection of actin isoforms in fixed cells, immunolabeling provides more stable images and less background signal than the RLP procedure. However, both of these techniques were commonly successful only with isolated plant cells (e.g., Meijer and Simmonds 1989; White and Sack 1990), epidermal peels (Cho and Wick 1991), or hand sections of fresh tissue (Koropp and Volkmann 1994).
To study the actin cytoskeleton in plant tissues, a suitable embedding technique is needed to view the cells in their spatial context and to relate the cytoskeleton arrangement to the differentiation state of the cell. Investigators face several difficulties in this situation. The actin cytoskeleton was reported to be very sensitive to formaldehyde (FA) fixation (Parthasarathy et al. 1985), and the use of crosslinker n-maleimido-benzoic acid N-hydroxysuccinimide ester (MBS) was recommended to protect microfilaments (Sonobe and Shibaoka 1989). Paraffin embedding does not preserve fine microfilaments, at least in plant tissues (McLean et al. 1990), probably because of the high temperatures used during embedment. Cryosectioning (Kobayashi et al. 1994) is labor-intensive and usually results in poor structural preservation of the plant tissue as well as of the actin cytoskeleton. Embedding in methacrylate resins (Baskin et al. 1992) provides good structure, but the tissue antigenicity does not appear to be optimal.
In our previous work on microtubules (Baluŝka et al. 1992, 1995) we had success with embedding of plant tissues in low melting point ester wax (Steedman's wax) (Brown et al. 1989). This embedding medium allowed convenient handling and sectioning and the tissue antigenicity was also excellently preserved. We present a modification of this procedure that enabled us to visualize various F-actin arrays on sections taken from intact root and stem tissues of Zea mays and Lepidium sativum roots and incubated with a monoclonal antibody (MAb) raised against animal actin.
Materials and Methods
Seven-mm-long root tip segments of 3-day-old Zea mays L. cv. Alarik seedlings were used to test different variants of the procedure. Stem nodes from 6-week-old Z. mays L. cv. Alarik and 7-mm-long root tips of 2-day-old L. sativum seedlings were utilized to verify the optimized procedure.
Objects were optionally vacuum infiltrated with 100 μM MBS (100 mM stock solution in DMSO) in stabilizing buffer (SB; 50 mM PIPES, 5 mM MgSO4, 5 mM EGTA; pH set to 6.9 using KOH pellets) for 15–60 min and then transferred to fixative: (a) 3.7% w/v FA in SB; (b) 1.5% w/v FA in SB; (c) 3.7% w/v FA in SB containing 10% v/v DMSO; (d) 0.5% w/v ZnCl2 in SB. The fixation with a, b, and c was done at room temperature (RT) for 30 min to 72 hr. Fixation with d and a was performed for 10 min at RT, then for 1 min in a microwave oven, which increased the temperature to 60C, and then the samples were transferred to the same fixative at RT for 10 min. After three washes of 10 min each in SB, and two washes of 15 min each in PBS (0.14 M NaCl, 2.7 mM KCl, 6.5 mM Na2HPO4, 5 mM KH2PO4, 3.0 mM NaN3; pH 7.3), the specimens were dehydrated in a graded ethanol/PBS series (30, 50, 70, 90, 97, 100% ethanol), each step for 30 min. For dehydration and embedding, analytical grade ethanol was used. The embedding medium was prepared in advance by melting 900 g polyethylene glycol 400 distearate at 60C and adding 100 g of 1-hexadecanol (both from Aldrich; Milwaukee, WI) and stirring for several hours. The wax was then stored at RT. Embedding was done at 35–37C, first for 30 min in 100% ethanol, then in graded wax/ethanol series (1+2, 1+1, 2+1 v/v) followed by three changes of pure wax, each for 1–2 hr. Objects were then put into embedding molds and left to polymerize overnight at RT.
Ribbons of 7-μm sections were placed on poly-L-lysine-coated slides and stretched by addition of a small drop of H2O to one end of the ribbon. The excess water was soaked away from the opposite end of the ribbon by filter paper. Slides were allowed to dry overnight at RT. For dewaxing and rehydration of sections, technical grade ethanol (Rotisol; Carl Roth, Karlsruhe, Germany) was employed containing 5% v/v acetone and 1% v/v methylethylketone. After dewaxing three times for 10 min in ethanol and rehydration in ethanol/PBS for 10 min each step (90%, 50% v/v, PBS alone), the sections were finally left for 30 min in SB. Optionally, the slides were then dipped in absolute methanol precooled to −20C or at RT. The 100-ml staining jar with methanol and slides was then placed in a −20C freezer for 10 min. Methanol treatment was followed by a 30-min SB wash. On some sections, hemicellulase digestion and Triton treatment were performed to improve the penetration of the antibodies. Sections were incubated for 20 min with 1% w/v crude hemicellulase (Sigma Chemical; St Louis, MO) in SB containing 0.5 mM EGTA, 0.4 M mannitol, 1% Triton X-100, and 0.3 mM phenylmethylsulfonyl fluoride. Sections were washed first with SB for 20 min, then in 1% Triton X-100 in SB for 10 min, and finally in SB for 20 min.
Sections were incubated with a mouse anti-actin MAb, clone C4 (Lessard 1988), purchased from ICN Pharmaceuticals (Costa Mesa, CA) diluted 1:200 or 1:400 in PBS, containing 0.1% w/v bovine serum albumin and 0.1% w/v NaN3. Two types of negative controls were performed: (a) the primary antibody was omitted or (b) IgG from normal mouse serum was used instead of the MAb at a corresponding protein concentration. Incubation was done in a moist chamber for 90 min at RT or at 37C. Slides were then washed twice for 10 min in SB and incubated for 90 min with secondary antibody, goat anti-mouse IgG-FITC conjugate (Sigma) diluted 1:100 in the same way as the first antibody. Incubation was performed at the same temperature as with the first antibody (RT or 37C) in darkness. Slides were then washed for 10 min in PBS. The nuclei were fluorescently stained with 4,6-diamidino-2-phenylidone (DAPI, 1 mM in PBS) for 10 min, followed by a 10-min wash in PBS. Sections were then stained for 10 min in 0.01% w/v Toluidine blue dissolved in PBS to suppress the autofluorescence of the cell walls (Brown et al. 1989). After washing for 10 min in PBS, the sections were mounted under a coverslip using p-phenylenediamine anti-fade mountant (Krenik et al. 1989).
Detection of F-actin by RLP (Molecular Probes; Eugene, OR) was performed as described by Tewinkel et al. (1989) on sections after dewaxing and/or optional treatment with cold methanol (see above).
Fluorescence was examined with an Axiovert 405 M inverted microscope (Zeiss; Oberkochen, Germany) equipped with epifluorescence and appropriate filter sets. Photographs were taken on Kodak T-max film 400 ASA.
The quantitative evaluation of the effect of fixatives used was performed by calculating percent of cells with actin microfilaments preserved. Five photomicrographs corresponding to that of Figure 1E were assessed for each variant. Oneway analysis of variance and Newman-Keuls multiple comparisons were performed using WinKS statistical software (TexaSoft; Cedar Hill, TX).
Results
The immunofluorescence procedure enabled us to visualize actin microfilaments in cells of various developmental stages in the plant tissues studied. Table 1 shows the effect of various fixatives after MBS pretreatment. Neither FA fixative containing 10% DMSO nor FA with microwave treatment preserved microfilaments. The best results were obtained with FA in SB at room temperature and with zinc chloride with microwave irradiation (Table 1). Zinc chloride-fixed roots gave stronger staining in the root cap and promeristem compared to the FA-fixed material. In both variants, however, the staining pattern in root cap cells was not filamentous (Figures 1C and 1D). FA was preferred in further experiments, because actin microfilaments appeared to be more intact. MBS-pretreated (15 min) specimens gave identical results after 30 min or 72 hr of FA fixation. No gross differences were found among FA- or zinc-fixed specimens when MBS pretreatment ranged from 15 to 60 min. The only exception was labeling of the phragmoplasts, which was successful only if MBS treatment did not exceed 15 min. Omission of MBS pretreatment had no dramatic effect when it was followed by FA fixation (60 min; data not shown). Zinc fixation without MBS pretreatment led to significant damage of microfilaments.
Fixation variants tested for actin immunofluorescence in Zea mays root tips
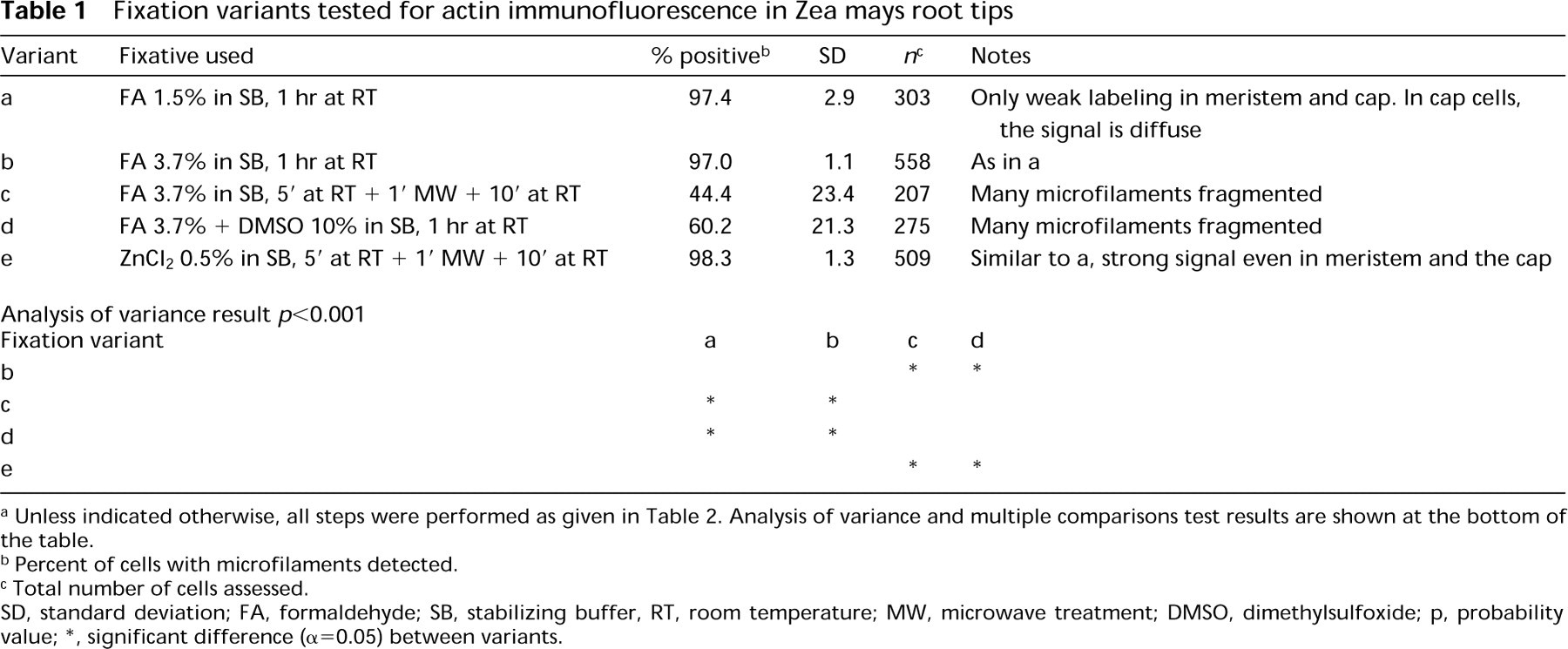
a Unless indicated otherwise, all steps were performed as given in Table 2. Analysis of variance and multiple comparisons test results are shown at the bottom of the table.
b Percent of cells with microfilaments detected.
c Total number of cells assessed.
SD, standard deviation; FA, formaldehyde; SB, stabilizing buffer, RT, room temperature; MW, microwave treatment; DMSO, dimethylsulfoxide; p, probability value; ∗, significant difference (α=0.05) between variants.
Hemicellulase digestion and Triton X-100 extraction caused fragmentation or even total destruction of microfilaments. Incubation with antibodies at RT led to better preservation of F-actin than incubation at 37C.
Methanol treatment was not necessary for good results. Without the use of methanol the signal was much stronger, but the background fluorescence was higher as well. Best results were obtained when sections were submerged in methanol at RT and placed for 10 min in a − 20C freezer.
First antibody diluted 1:200 gave optimal results. Controls in which the sections were incubated in the presence of preimmune IgG (Figure 1A) or in which the primary anti-actin MAb was omitted did not show any staining above faint diffuse background. The optimized procedure is summarized in Table 2 and its results are shown in Figures 1 and 2. Actin microfilaments were more abundant in cells of the stele than of the cortex (Figures 1E and 2B). Only faint signal was observed in cells of the quiescent center (Figure 1B). In interphase cells of the root cortex, cytoplasmic and cortical MFs were visualized (Figures 2B-2D). In postmitotic cells of the stele, bundles of MFs formed a cone-shaped cage around the nucleus and stretched towards the end walls (Figures 1E, 1F, and 2A). A similar pattern was also seen in postmitotic cortex cells, although not as pronounced.
All attempts to visualize actin microfilaments by the RLP technique failed in both methanol-treated and untreated sections.
Discussion
The procedure introduced here allows, for the first time, easy and sensitive detection of extensive networks and fine bundles of actin filaments on serial microtome sections of plant tissues incubated with an anti-actin MAb. The embedding medium, low melting point (35C) ester wax, allowed gentle embedding at low temperature (35–37C), which is only about 15C higher than the cultivation temperatures common for most plants. The preservation of antigenicity in this medium is excellent, allowing short incubation times. The physical characteristics of the medium are similar to those of paraffin, and it is easy to obtain ribbons with serial sections. Because of the low melting point of the wax, in hot weather it might be necessary to perform the microtomy in a cold room and to cool the knife in a refrigerator. The use of a rotary microtome and high cutting speeds is recommended for best results. Sections 4–20 μm thick can be routinely obtained, which enables one to adjust the section thickness to the average cell size for a given tissue.

Immunofluorescence of actin microfilaments in maize root cells. Procedure as described in Table 2 except for D, in which ZnCl2 fixation was used (see Materials and Methods).
Optimized immunofluorescence procedure for F-actin visualization a
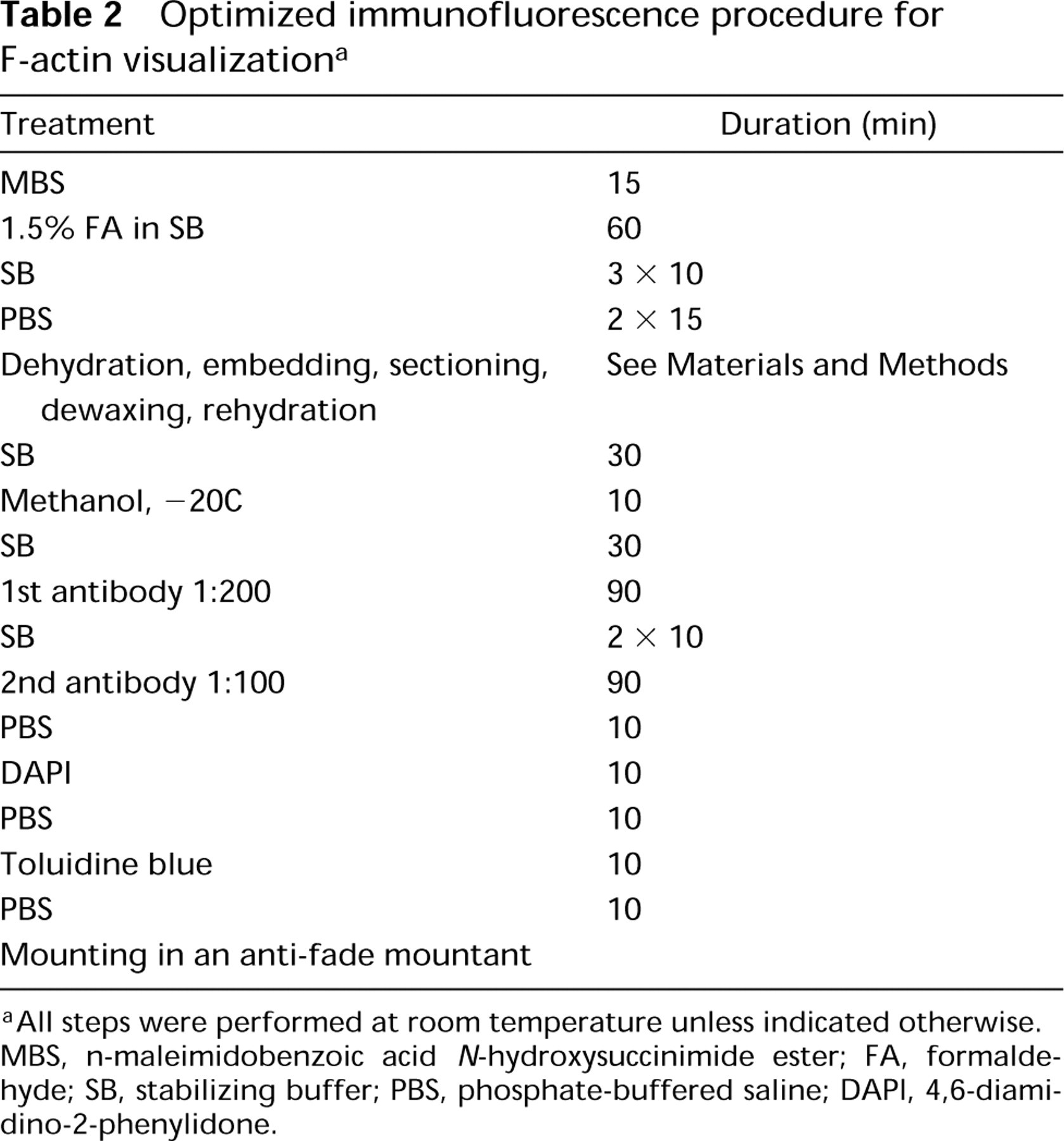
a All steps were performed at room temperature unless indicated otherwise. MBS, n-maleimidobenzoic acid N-hydroxysuccinimide ester; FA, formaldehyde; SB, stabilizing buffer; PBS, phosphate-buffered saline; DAPI, 4,6-diamidino-2-phenylidone.
The conclusion that the staining patterns observed are due to presence of actin is based on the following. (a) The antibody used (C4) was shown to bind to an antigenic determinant highly conserved among the actins (Lessard 1988). (b) Cho and Wick (1991) found the same staining patterns when they compared C4 (ICN Pharmaceuticals) and N350 (Amersham; Arlington Heights, IL) antibodies and rhodamine-phalloidin staining in rye. (c) The intracellular arrangement of actin visualized in the presented procedure by the C4 antibody is similar to that detected by other antibodies or rhodamine-phalloidin in plant cells and tissues (Meijer and Simmonds 1989; Vaughan and Vaughn 1987). (d) The negative controls with normal IgG instead of the anti-actin MAb did not show any filamentous staining pattern (see Figure 1A). For immuno-blotting with C4 and N350 antibodies, see Koropp and Volkmann (1994).
The positive effect of MBS pretreatment on F-actin preservation is in agreement with the data of Sonobe and Shibaoka (1989). Nevertheless, the omission of MBS did not lead to substantially worse results in our material. On the other hand, it appears that different microfilament arrays are affected differently by MBS. This is illustrated by the inability to detect phragmoplasts if MBS pretreatment exceeds 15 min. More data from other plant species and organs are needed to evaluate the effect of MBS in the procedure. At present, the use of MBS is recommended. Zinc chloride fixation gave a stronger signal in the promeristem and root cap, where the use of FA fixation led to only a weak signal. This is in accord with the original report of Beckstead (1994), who found zinc fixatives superior to FA for preserving antigenicity in paraffin-embedded animal tissues. However, the effect of zinc chloride on the plant tissue has not been thoroughly characterized. It is not known how rapidly the cells are killed and the biochemical processes stopped by the zinc chloride fixation used in our experiments. Microwave irradiation raised the temperature very quickly to 60C (within 1 min) and extensive postfixation changes therefore appear to be unlikely. The possibility of such artifacts, however, cannot be completely ruled out. We therefore adhered to FA fixatives, which are routinely used in many microscopic procedures.
Intensive labeling of actin microfilaments could be achieved without treating the sections in methanol. This may appear to be in contrast with the findings of Cho and Wick (1991), who concluded that anti-actin antibodies react only with actin treated with organic solvent, such as acetone or methanol. It should be stressed, however, that the ethanol used for dewaxing our sections was of technical grade (chosen for its low cost) and contained 5% v/v acetone (see Materials and Methods).
Surprisingly, incubation with antibodies at RT gave remarkably better results (both in intensity and in F-actin preservation) than that at 37C. It follows that the microfilament arrays in the sectioned material, although it has been fixed, are extremely sensitive to high temperature and are still subject to damage by incubation conditions. Actin microfilaments were detected in all growing cells of the root proper, which indicates good penetration of the antibodies in 7-μm sections of the root tip. Differences in relative fluorescence intensities within the section (stele vs cortex) or absence of fluorescence (quiescent center) can be explained by differences in F-actin abundance rather than by differential penetration of antibodies. Given the size of the cells, all of them were probably cut by the microtome knife, and antibody penetration therefore does not appear to be a problem. These differences are also unlikely to be solely due to unequal preservation of microfilaments. As shown in Figure 2C, fine actin microfilaments are preserved in both stele and cortex cells.
The present method appears to be incompatible with detection of F-actin by RLP. Therefore, direct comparison of these two methods is not possible here. Although the RLP technique is often believed to provide better preservation of fine actin microfilaments, Schmit and Lambert (1987) obtained better resolution with immunofluorescence. Walker and Sack (1995) found less extensive staining of F-actin with anti-actin antibody than with RLP. The results of the former technique were explained as an artifact caused by fixation. It is possible, however, that polymerization of actin in the presence of RLP might have contributed to these differences.
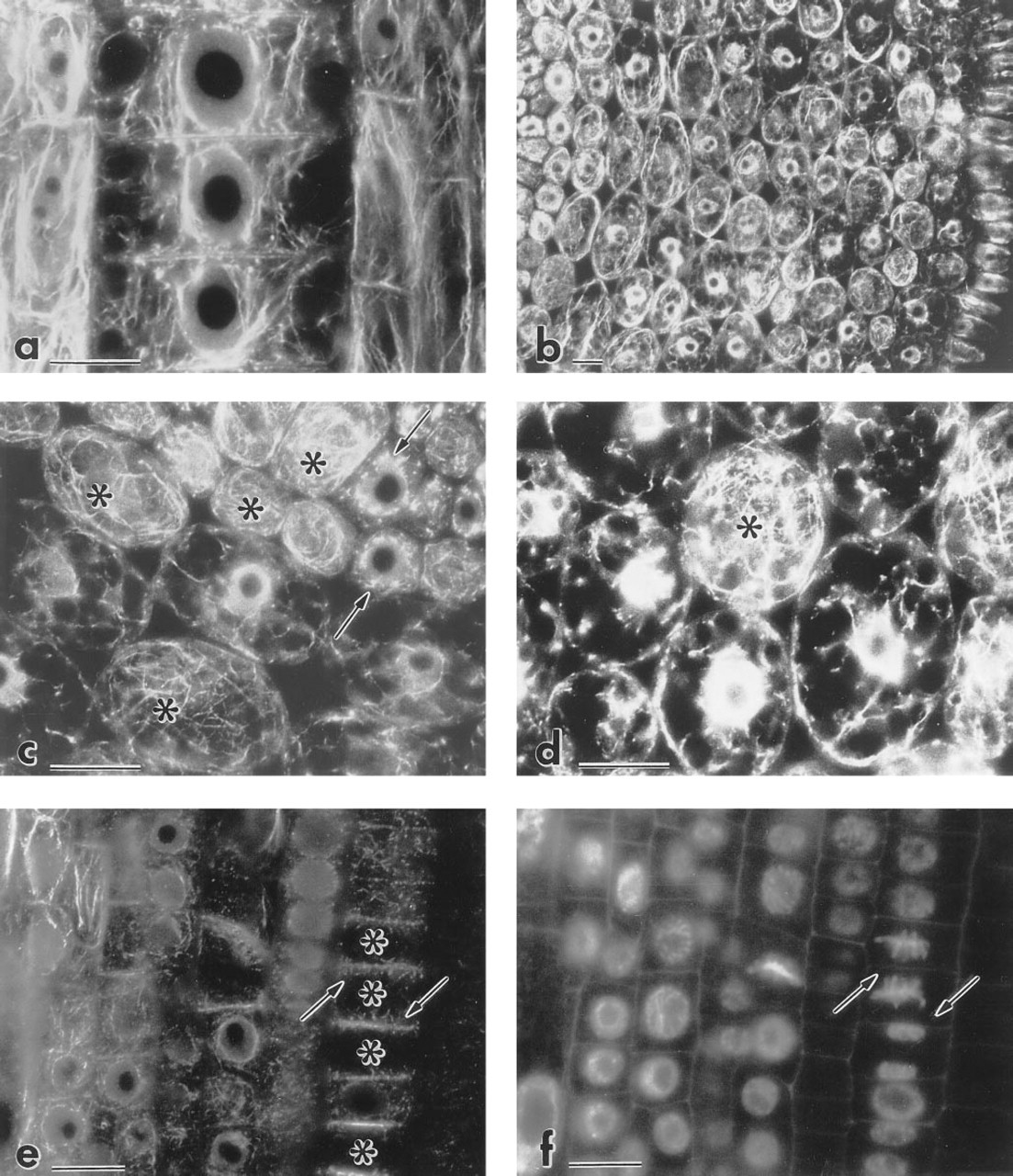
F-actin immunofluorescence in maize root tips. Procedure as described in Table 2.
The procedure presented here is a contribution to the tools available for the study of actin microfilaments, because it allows actin filaments to be visualized in sections from intact plant tissues. In this way, developmental changes in the cytoskeleton can be documented.
Footnotes
Acknowledgements
SV was supported by a Deutsches Akademisches Austauschdienst (DAAD; Bonn, Germany) fellowship. FB was supported by a fellowship from the Alexander von Humboldt Foundation (Bonn). Financial support to AGRAVIS (Bonn) by the Deutsche Agentur für Raumfahrtangelegenheiten (DARA, Bonn) and the Ministerium für Wissenschaft und Forschung (MWF, Düsseldorf) is gratefully acknowledged.