
Abstract
Immunogold labeling of ultrathin cryosections provides a sensitive and quantitative method to localize proteins at the ultrastructural level. An obligatory step in the routine preparation of cryosections from cultured cells is the detachment of cells from their substrate and subsequent pelleting. This procedure precludes visualization of cells in their in situ orientation and hampers the study of polarized cells. Here we describe a method to sample cultured cells from a petri dish or coverslip by embedding them in a 12% gelatin slab. Subsequently, sections can be prepared in parallel or perpendicular to the plane of growth. Our method extends the cryosectioning technique to applications in studying polarized cells and correlative light–electron microscopy.
C
The immunogold labeling technique can be combined with various preparation techniques. In preembedding protocols, cell membranes are made permeable to the immunoreagents by chemical or mechanical treatment. These approaches usually result in high labeling densities, but permeabilization may lead to a severe loss of fine structural integrity. Permeabilization is avoided in postembedding techniques, in which immunoreactions are performed on the surface of ultrathin sections of plastic- or Lowicryl-embedded cells or tissues. A drawback of postembedding methods, however, is that the antigenicity of molecules is often severely affected by the harsh treatment with organic solvents and dehydration of the specimen necessary for embedding.
The disadvantages of the pre- and postembedding techniques are avoided by ultracryotomy, a method by which ultrathin sections are prepared from non-permeabilized, non-embedded mildly fixed cells or tissue (Tokuyasu 1973,1978,1980; Griffiths 1993). Sections are prepared at low temperature, which yields the necessary support and hardness for sectioning. The frozen sections are removed from the cryoultramicrotome, thawed at room temperature (RT) in a watery environment, subjected to the immunolabeling procedure, and only then embedded in a thin layer of methylcellulose/uranyl and air-dried. The methylcellulose/uranyl step prevents drying artifacts and provides the necessary membrane contrast and support. The cryosectioning technique is usually several times more sensitive than postembedding methods. In addition, cryosections show excellent membrane preservation, especially when the increased methylcellulose/sucrose pick-up method developed by Liou et al. (1996) is applied. Hence, immunogold labeling of ultrathin cryosections provides a sensitive and quantitative tool, which has been widely used to detect proteins involved in normal and pathological cell functioning.
The preparation of cryosections of cultured cells involves a mild fixation, after which cells are removed from the carrier substrate with a cell scraper or enzymatic proteolysis and subjected to pelleting (Griffiths 1993; Kleijmeer et al. 1996; Raposo et al. 1997). Sections through the pellet contain random cell profiles, which precludes the study of cells in their natural orientation. We experienced this as a limitation of the cryosectioning technique when we were studying axon transport in polarized neurons, which required cutting cells in parallel to the substratum without destroying contact sites or membrane continuities. We therefore developed a method by which cryosections can be made in parallel, or perpendicular, to the plane in which cultured cells are grown. As model systems we used non-differentiated neuroendocrine PC12 cells and primary cultures of mouse hippocampal neurons (Annaert et al. 1999). Our method extends the general applicability of the cryosectioning technique to polarized cells and correlative light–electron microscopy (CLEM).
Materials and Methods
Antibodies
Rabbit antisera against vesicle-associated membrane protein-2 (VAMP-2) and synaptophysin, and mouse monoclonal antibody against rab3 A/B/C/D (Cl 42.1; Matteoli et al. 1991) were obtained from Synaptic Systems (Göttingen, Germany) and used at a dilution of 1:25, 1:100, and 1:500, respectively. A monoclonal antibody (MAb) against clathrin heavy chain was obtained from BD Transduction Laboratories (San Diego, CA) and used in a dilution of 1:100. Immunogold labeling was performed as described previously (Slot et al. 1991), using protein A conjugated to 10-nm gold particles, with the minor modification that antibodies were diluted in PBS containing 0.5% fish skin gelatin (Sigma-Aldrich; Zwijndrecht, The Netherlands) and 0.1% acetylated bovine serum albumin (Aurion; Wageningen, The Netherlands). When a mouse MAb was used as the primary step, a bridging rabbit anti-mouse IgG antibody (DAKO; Glostrup, Denmark) was applied at a dilution of 1:250 to provide binding sites for protein A–gold. For immunofluorescence (Figures 7 and 8A), mouse MAb Cl69.1 against VAMP-2 (Synaptic Systems) was used at a dilution of 1:1000.
Culture of PC12 Cells
PC12 cells (clone 251-II) were routinely grown on 35-mm petri dishes in Dulbecco's modified Eagle's medium supplemented with 10% horse serum and 5% fetal calf serum at 37C under 10% CO2 (Greene and Tischler 1976; de Wit et al. 1999).
Low-density Cultures of Hippocampal Neurons from Mouse Brain
When grown at low density, hippocampal neurons go through five clearly defined developmental stages that closely resemble the in vivo situation (Dotti et al. 1988; Goslin and Banker 1991). At the first day of culture, cells form flattened lamellipodia (stage 1) that rapidly condense to short minor processes (stage 2). One of these processes elongates and acquires axonal properties (stage 3, day 1.5). After 3–4 days, the minor processes begin to elongate and specialize into dendritic processes (stage 4). In subsequent days, the density of the axon network increases and arborization of dendrites becomes prominent. At this fifth stage (day 7 and beyond) hippocampal neurons develop synaptic contacts and dendritic spines in large numbers (Annaert et al. 1999). In this study only fully differentiated and polarized murine hippocampal neurons were used, fixed at day 14 after plating in 2% paraformaldehyde and 0.2% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4. To obtain a differentiated pheno-type, the neurons must be cultured on a feeder layer of glial cells. Monolayers of glial cultures were prepared as described by Goslin and Banker (1991). Briefly, the cerebral hemispheres of three newborn mice were dissected and minced with scissors. After trypsinization [0.25% trypsin in Hanks' Balanced Salt Solution (HBSS)] for 15 min at 37C, cells were plated on poly-
Immunofluorescence
Hippocampal neurons grown for 14 days on coverslips were fixed with 4% paraformaldehyde in 0.1 M phosphate buffer overnight at 4C. Fixative was removed by three washes in PBS. After quenching free aldehyde groups with 50 mM NH4Cl in PBS for 5 min and an additional wash in PBS, the cells were permeabilized for 1 hr in 0.5% BSA in PBS containing 0.1% saponin (blocking solution). The BSA in this solution reduces nonspecific protein–protein interactions of the antibodies. Cells were incubated with an MAb against VAMP-2 for 30 min and then washed three times for 5 min with blocking solution. A Cy3- (Jackson Laboratories; West Grove, PA) or FITC- (DAKO; Glostrup, Denmark) conjugated rabbit anti-mouse IgG secondary antibody was diluted in blocking solution and applied for the next 30 min. After several washes, cells were mounted in Mowiol on an object slide and viewed with a x63 planapo objective on a Leitz DMIRB fluorescence microscope (Leica; Voorburg, The Netherlands) interfaced with a Leica TCS4D confocal laser scanning microscope (Figure 7), or on a Leica Polyvar epifluorescence microscope (Figure 8A).
Standard Procedure for Preparation of Ultrathin Cryosections from Cultured Cells
Cryosections from cells grown on a solid substrate can be prepared using various adaptations of the standard protocol developed by Tokuyasu (1973). To understand the flat-embedding procedure for ultrathin cryosectioning described in this paper it is necessary to know the standard procedure, which we therefore briefly describe here (for recent reviews and more detailed protocols see Kleijmeer et al. 1996; Raposo et al. 1997).
In general, cells are fixed at a minimum for 2 hr at room temperature (RT) or overnight in the cold. The fixative is rinsed away by washing twice with PBS to which 0.02 M glycine is added to quench free aldehyde groups. A small volume of 1% gelatin (Twee Torens; Delft, The Netherlands) in PBS at RT is then added, in which the cells are removed from the dish with a cell scraper. Cells are transferred to an Eppendorf vial and centrifuged for 30–60 sec at 1500 rpm, after which the 1% gelatin is replaced by a 12% gelatin solution at 37C. After resuspension, cells are either centrifuged again, after which the excess gelatin is removed (procedure A), or transferred to a microscope slide (procedure B). In the latter case, a small petri dish is placed, bottom down, on the top of the droplet to transform it into a slab. In both procedures the gelatin solution is solidified for 30 min on ice. The 12% gelatin provides the necessary support to handle the cells during the following preparation steps, which are all performed in the cold room.
In procedure A, the bottom of the Eppendorf tube is cut off with a razor blade, after which 1-mm3 gelatin blocks are prepared. Similar blocks can be directly prepared in procedure B. The small blocks are rotated in 2.3 M sucrose in phosphate buffer for at least 4 hr at 4C. Sucrose acts as a cryoprotectant, which abolishes ice crystal formation during the subsequent freezing step. After sucrose impregnation the blocks are placed on top of an aluminum specimen holder, the excess of sucrose is removed, and the specimen plus holder are quickly plunged into liquid nitrogen.
Sectioning begins with removal of the sucrose cover and flattening of the front of the specimen. The sides of the specimen are trimmed with a razor blade, the corners of a glass knife, or with a specialized trimming diamond. After trimming the specimen to an optimal rectangular shape, ultrathin sectioning with a diamond knife is performed at about −120C. Sections of 60–65-nm thicknesses are removed from the knife with a 1:1 mixture of 2% methylcellulose and 2.3 M sucrose (Liou et al. 1996) and transferred to a grid (copper grids bearing a carbon-coated Formvar supporting film). Sections can be stored at 4C (Griffith and Posthuma 2002) or directly subjected to the single or double immunogold labeling procedure as described (Slot et al. 1991). The labeling procedure is carried out at RT on thawed sections. On warming the grids to RT the gelatin melts. Gelatin therefore does not hamper incubation with immunoreagents. After completion of the labeling, sections are embedded in a thin layer of 2% methylcellulose with 0.4% uranyl acetate, pH 4.0, and air-dried.
This procedure results in the random visualization of cell profiles of pelleted cells. Examples of data obtained according to this procedure are numerous. For recent studies see, e.g., Mártinez–Menárguez et al. 1999,2001. In the paper by de Wit et al. (1999), non-polarized PC12 cells prepared according to this procedure are shown.
Results
Preparation of a Cell Slab from a Plastic Petri Dish
To overcome the necessity to make cell pellets and be able to section cells in parallel to the dish, we have developed a method that avoids scraping of the cells and allows removal of intact cells from a plastic petri dish (method A in Figure 1). We performed our initial studies with non-differentiated PC12 cells fixed after 7 days of culture. Non-differentiated PC12 cells are grown directly on the petri dish, but experiments with poly-
To enhance sucrose infiltration in the double-gelatin layer, the gelatin was scarified from top to bottom with a scalpel (Figure 1A2). Cells were then subjected to a prolonged infiltration with 2.3 M sucrose for 48–72 hr at 4C on a rocker. The sucrose infiltration, in addition to cryoprotection, caused a slight shrinking of the gelatin layers, resulting in loosening of the gelatin slab plus cells from the dish. Notably, because the gelatin melts when a section is transferred to RT, at which the labeling procedure is carried out, it does not interfere with the immunoreaction. Large pieces of the gelatin/cell slab could then be detached from the dish with a thin spatula (Figure 1A3). These blocks were stained for 30 min at 4C in a solution of 1% Toluidine blue to which 2.3 M sucrose was added to better preserve ultrastructure. The Toluidine blue staining is optional. We found that it allowed detection of cells in 1-μm cryosections that were viewed under the binocular and therefore enabled selection of areas of interest for further processing. Moreover, it facilitated ultrathin sectioning because the stained cells were clearly visible in the microtome. Finally, the Toluidine blue staining was used as a control to check whether cells had accidently remained attached to the petri dish during the flat-embedding procedure. After selecting specific areas under the binocular, approximately 1-mm3 blocks of the gelatin slab were cut out with a razor blade, mounted on aluminum pins (Figure 1C1), and frozen in liquid nitrogen. For sectioning in parallel to the dish, the erythrocyte layer was oriented towards the pin (Figure 1C1a). Alternatively, the erythrocyte layer can be placed sideways (Figure 1C1b), which is of use when, for example, polarized epithelia are sectioned. A light microscopic image of a 1-μm-thick cryosection, prepared in the orientation as shown in Figure 1C1b, is shown in Figure 2. For our goal, ultrathin cryosections were cut parallel to the monolayer, picked up in a 1:1 mixture of 2.3 M sucrose and 2% methylcellulose (Liou et al. 1996) and immunogold-labeled according to the protein A–gold method (Slot et al. 1991). From the very first section on, sections were retrieved from the microtome, transferred to a grid (Figure 1C3), rinsed for 1 min with distilled water, dried, and analyzed in the EM for the presence of cells (quick screening method). When material was found present, all subsequent sections were collected and stored on a series of grids until further processing (Griffith and Posthuma 2002).
The flat-embedding procedure resulted in a superb ultrastructural preservation of non-differentiated PC12 cells (Figures 3 and 4). Staining with Toluidine blue (optional) resulted in a denser appearance of the cells in the EM, especially the nucleus (Figures 3A and 3B) but had no effect on labeling densities (Figures 3C–3F). Although designed for polarized cells, the flat-embedding approach also proved to be of interest to view non-differentiated cells. As shown in Figure 4, in areas of close contact PC12 cells alternate large areas of flattened plasma membrane with regions displaying microvilli. This type of ultrastructural information is only poorly or incompletely visualized when cells are scraped from the dish.
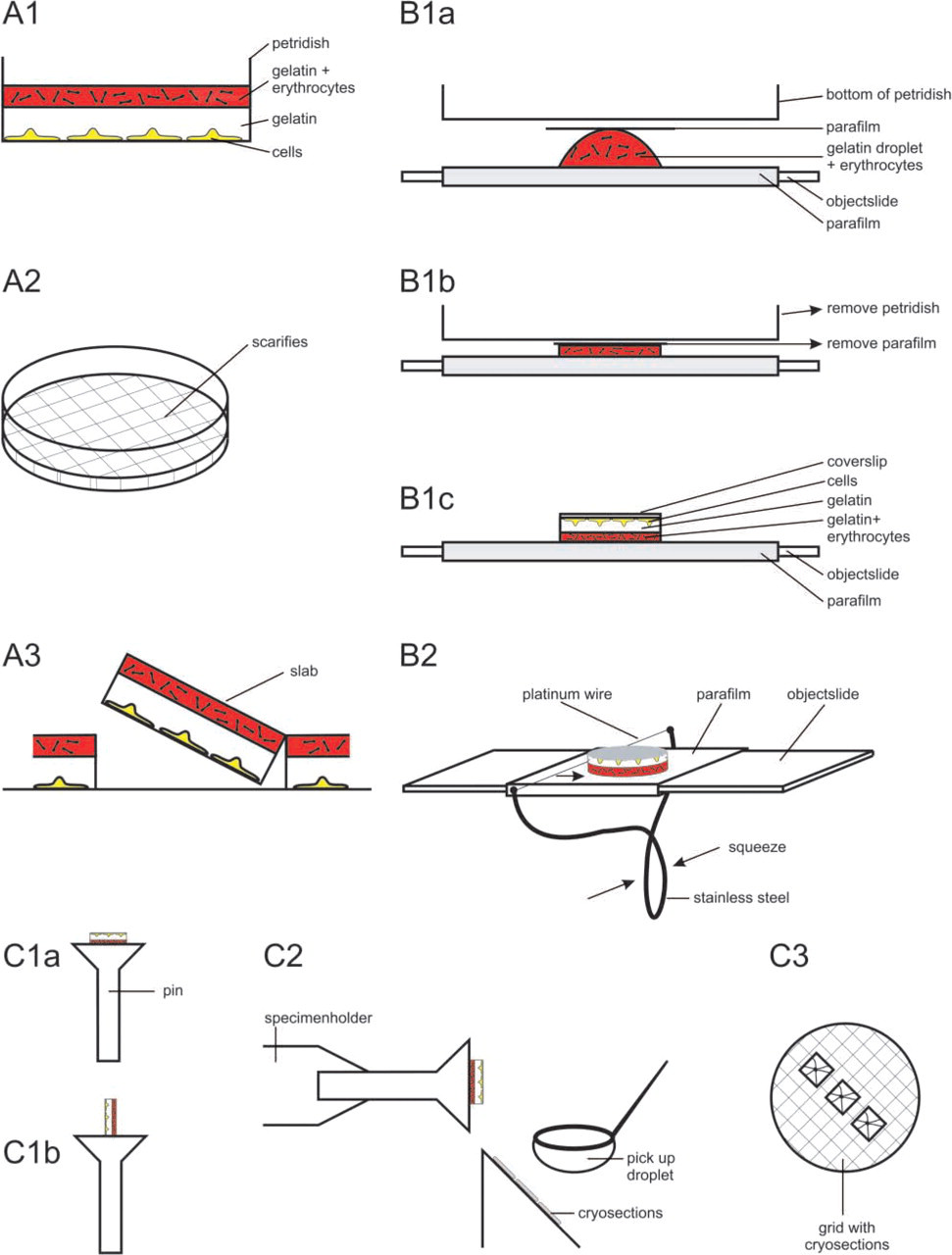
Schematic representation of the various steps required for the preparation of a gelatin slab from petri dishes (steps A1–A3) and coverslips (steps B1 and B2). Further steps are the same for cells grown on petri dishes and coverslips (C1–C3). The procedure is explained in detail in the Results section.

Light microscopic image of 1-μm-thick cryosection that was oriented at the pin as in Figure 1C1b. The cells at the left hand of the section are non-differentiated PC12 cells. Original magnification x33.
Preparation of a Cell Slab from a Glass Coverslip
Primary cultures of hippocampal neurons are an often used model system to study transport and development of polarized neuronal cells (Goslin and Banker 1991). Unlike PC12 cells, hippocampal neurons are grown on coverslips. To prepare cells from coverslips we slightly adapted the above-described method (method B in Figure 1). We used mouse-derived hippocampal neurons grown on glass or Thermanox coverslips, fixed in 2% formaldehyde and 0.2% glutaraldehyde, and rinsed in PBS just before flat-embedding. First, a glass object slide was tightly wrapped with parafilm (the parafilm was found necessary to detach the gelatin slab at the end of the procedure). On top of the parafilm a 70–100-μl droplet of a 37C solution of 12% gelatin with fixed erythrocytes was placed (Figure 1B1a). After 20 sec at RT, to let the gelatin solidify slightly, a small piece of parafilm was put on the droplet on which a 3.5-cm petri dish, bottom down, was placed to produce a flattened slab (Figure 1B1b). After 30 min solidification at 4C, the petri dish and small piece of parafilm were carefully removed from the gelatin–erythrocyte slab. Then the object slide with the gelatin–erythrocyte slab was incubated for 3 min at 37C and, when the gelatin–erythrocyte slab had melted slightly, a droplet of 37C 12% gelatin in PBS was situated on top of it. After 20 sec the coverslip with cells downward was placed on the 12% gelatin droplet, which transformed the gelatin into a second slab (Figure 1B1c). After solidification for 30 min at 4C, the coverslip with the entire layer of gelatin/neurons–gelatin/erythrocytes was detached from the object slide by pulling a platinum wire in between the parafilm and the gelatin/erythrocyte slab (Figure 1B2). In this step, it is important to get a flat surface of the gelatin/erythrocyte slab because irregularities will obstruct the sectioning of larger surfaces in the cryoultramicrotome. Therefore a special device was developed (see Figure 1B2), which enabled us to pull the platinum wire straight when dragged over the object slide. To further facilitate the detachment of the gelatin block from the object slide, a piece of parafilm that was slightly larger than the coverslip was cut out with a scalpel. With the parafilm on top, this was transferred to a 2.3 M sucrose solution pre-cooled to 4C and incubated for 4 hr on a rocker. Then the parafilm was carefully removed and the block was infused with sucrose for another 72 hr. The subsequent sucrose-induced shrinking of the gelatin resulted in loosening of the gelatin block from the coverslip, which then could easily be detached. Further preparation was as described in PC12 cells grown on a petri dish (Figures 1C1–1C3).
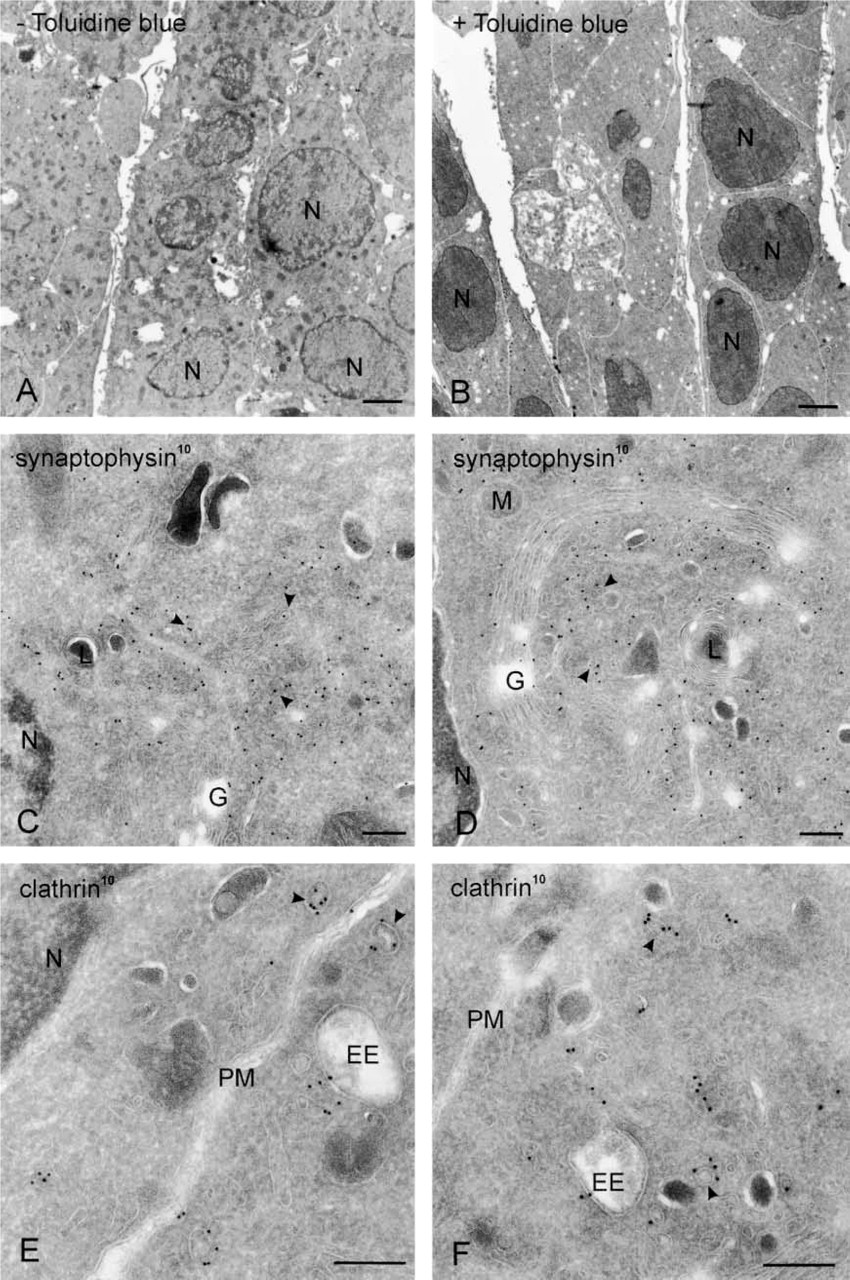
Cryosections of non-differentiated PC12 cells cut in parallel to the culture dish and prepared according to the flat-embedding procedure in the absence (
Cryosections of non-differentiated PC12 cells cut in parallel to the culture dish, prepared according to the flat-embedding procedure and immunolabeled for VAMP-2 (10-nm gold). (
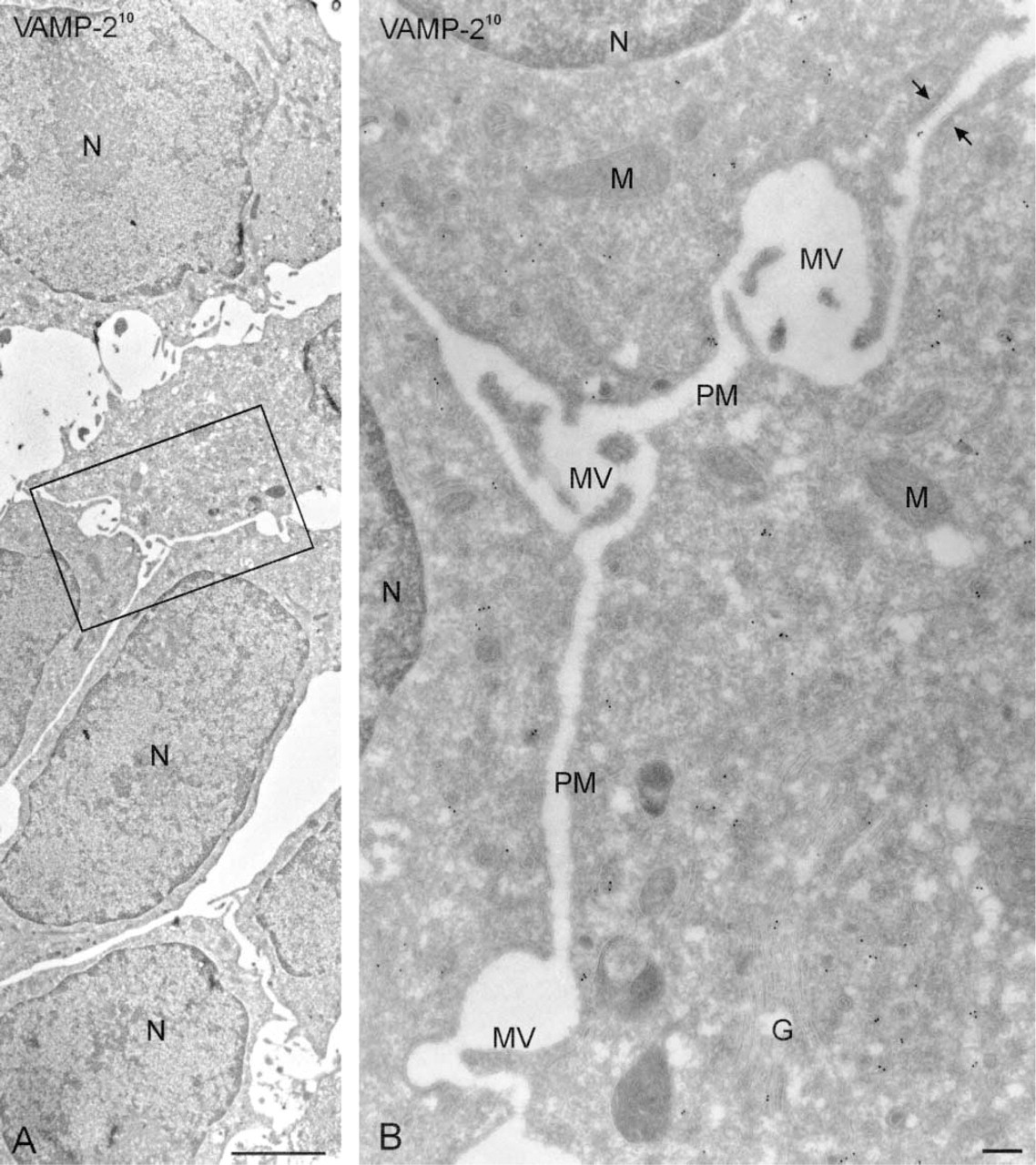
Because the neuronal extensions are directly opposed to the cutting edge of the knife (Figure 1C2) and have a limited height, they are present in the first 20–30 sections only. Therefore, blocks were not trimmed at the front. Ultrathin cryosections of hippocampal neurons embedded according to this procedure revealed a network of dendritic and axonal extensions emerging from the cell body (Figure 5). Immunogold labeling clearly provided important information to understanding of the complex architecture and the distinct membrane domains of the neuron. For example, immunogold labeling for rab3 revealed that some of the membrane-bounded structures close to the cell body contained high levels of rab3-positive synaptic vesicles (see Figure 5 inset), suggesting that these are cross-sections of synapses contacting the cell body. Immunogold labeling also discriminated axonal from dendritic extensions (Figure 6A). Axons but not dendrites contained high levels of rab3 (not shown) and VAMP-2 (Figure 6A). Many contact sites between axons and dendrites were observed, sometimes with the synaptic specializations clearly visible (Figure 6C). Synapses had a bulb-like (Figure 6B) or elongated (Figure 6C) shape.
CLEM
An important additional advantage of the flat-embedding technique is the possibility of performing CLEM. Staining of hippocampal neurons with VAMP2 by immunofluorescence results in a patchy staining pattern (Figure 7). Often two fluorescent tracks are running in parallel, delineating an unstained area (see also Figure 7 inset). With the information obtained in the EM (Figure 6), this peculiar staining pattern could be explained as a multitude of bulb-like and flattened synaptic contact sites outlining the postsynaptic dendrite. Although informative, this type of CLEM is indirect. For direct CLEM we subjected fluorescence-stained cells to the flat-embedding procedure. First, cells were stained for VAMP-2 by the routine fluorescence procedure described in the methods section, using an FITC-conjugated secondary antibody (Figure 8A). After photographing areas of interest in the fluorescence microscope, the coverslip with cells was removed from the object slide by rinsing in PBS, after which the procedure was carried out as described above. Areas selected under the fluorescence microscope were backtracked under the binocular, which was feasible because of the Toluidine blue staining. Cryosections of the selected areas were then made using the flat-embedding procedure and sections were labeled with anti-FITC antibody and 10-nm gold. Figure 8 shows a sequence of pictures of increasing magnification, illustrating how a fluorescent spot of VAMP2 (arrow in Figure 8A) is identified as a synaptic terminal in the electron microscope (arrows in Figures 8B–8D). At high magnification, it becomes clear that the gold particles indicating the presence of VAMP-2 are associated with the small synaptic vesicles in the synaptic terminal (Figure 8E). Equivalent data were obtained when an Alexa 488-conjugated secondary antibody was used for immunofluorescence and an anti-Alexa antibody on the ultrathin cryosections (not shown). This CLEM procedure resulted in good overall labeling density but the overall morphology at the ultrastructural level decreased compared to flat-embedded cryosections not previously stained for immunofluorescence, probably because of the saponin permeabilization step in the fluorescence procedure. Permeabilization can be avoided when proteins are made fluorescent by recombinant tagging with GFP or its derivatives and using an anti-GFP antibody for labeling of the cryosections. Finally, when serial sections are collected, multiple cross-sections of the structures previously identified by immunofluorescence can be examined, yielding possible additional information (not shown).
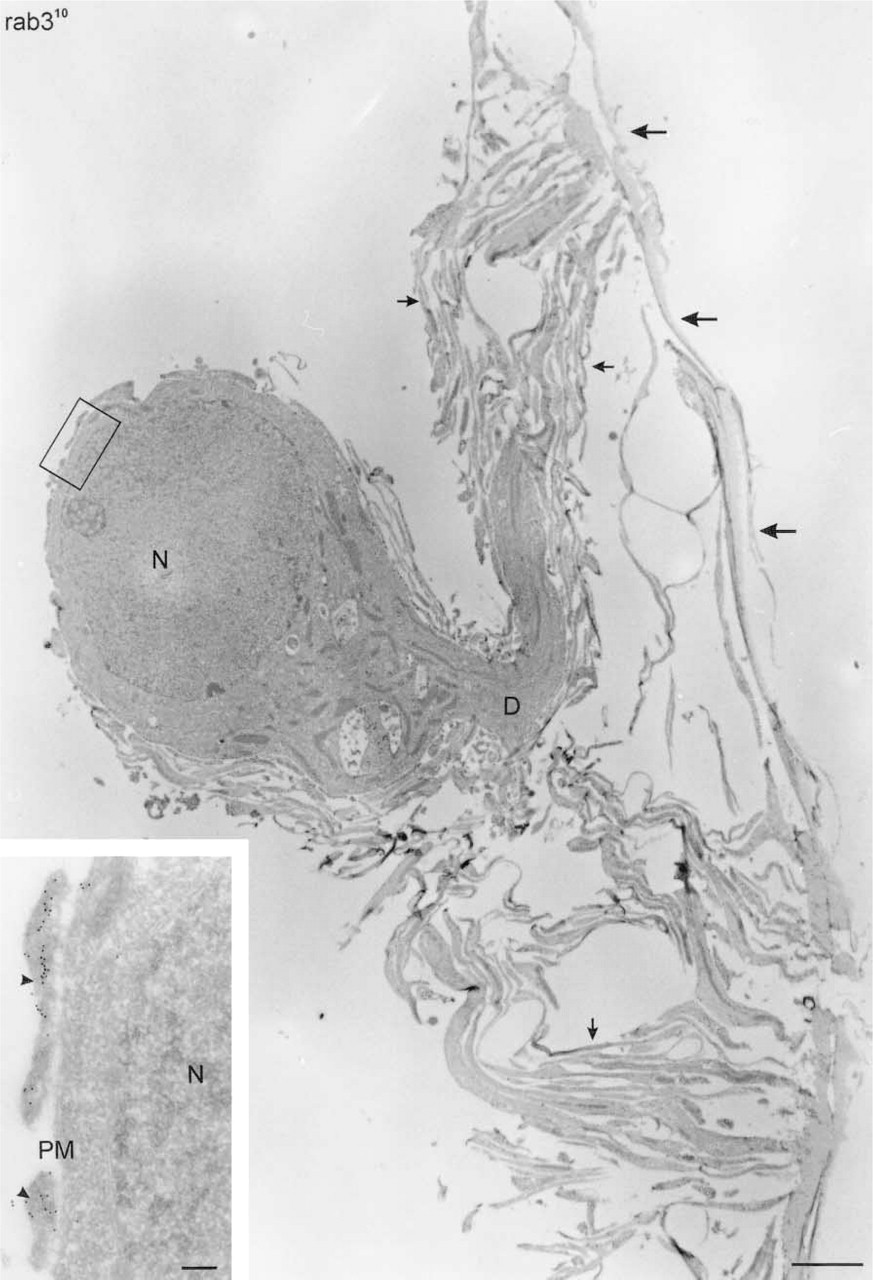
Ultrathin cryosection showing a low-magnification overview of a mouse hippocampal neuron, cultured for 14 days on a glass coverslip, cut in parallel to the coverslip, prepared according to the flat-embedding procedure, and immunolabeled for rab3. A thick dendrite (D) evolves from the cell body and is surrounded by many branches of dendritic and axonal origin (small arrows). Large arrows point to coat material that is sometimes removed from the coverslip during the procedure. Bar = 2 μm. (
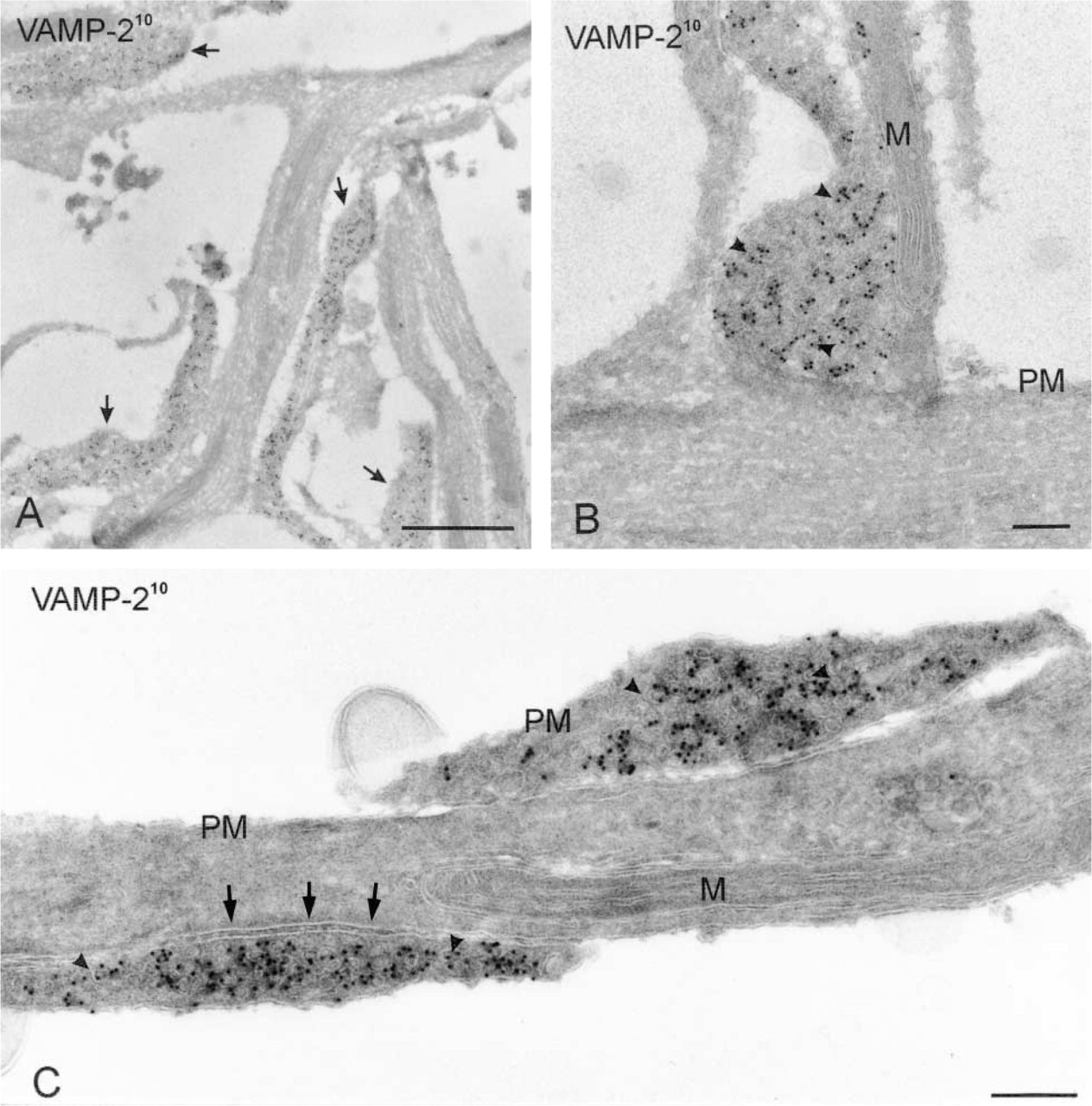
Cryosections of flat-embedded hippocampal neurites cut in parallel to the coverslip on which they were grown, immunogold labeled for VAMP2 (10-nm gold). (
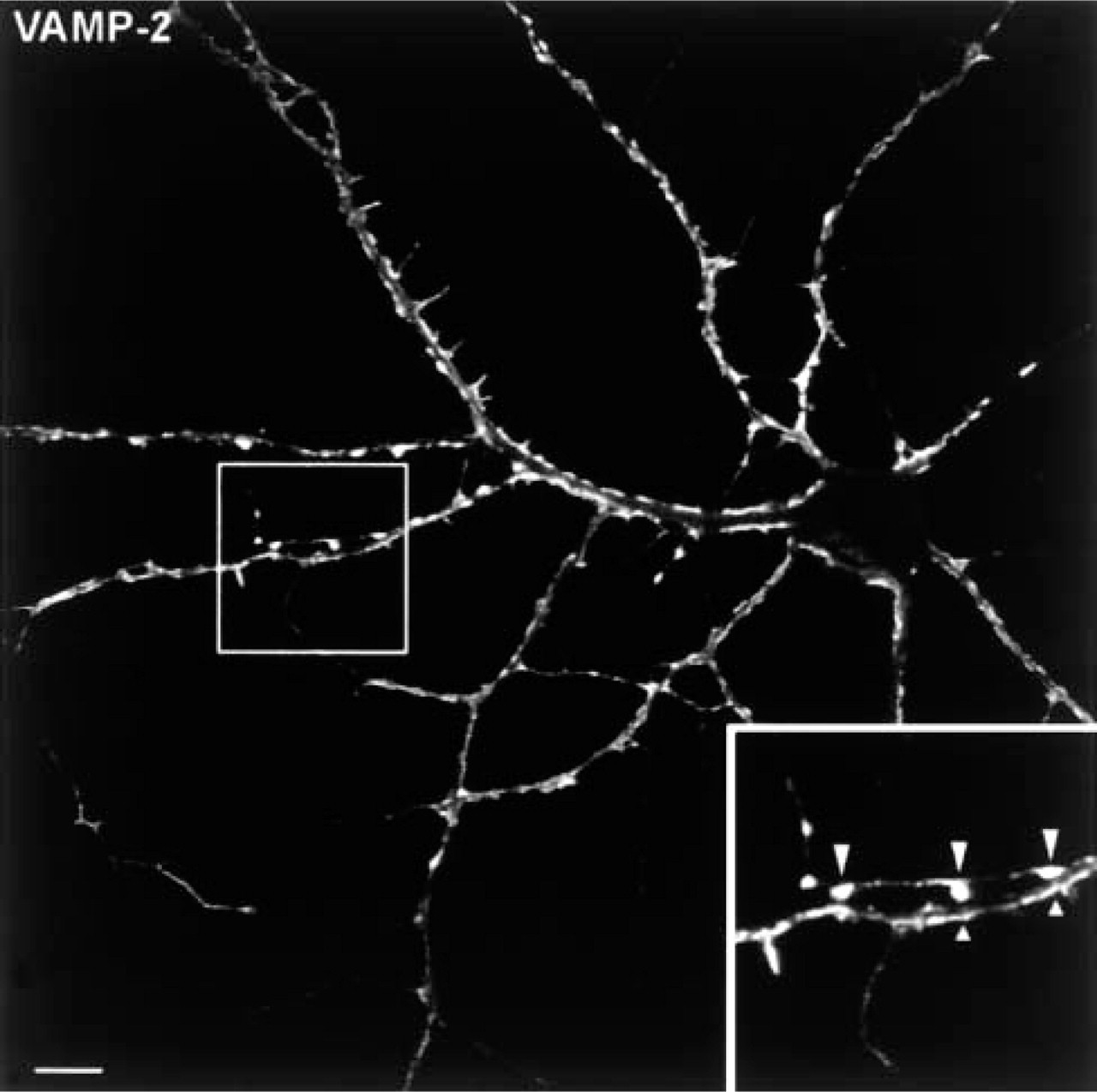
Immunofluorescence image of a hippocampal neuron stained for VAMP-2. The fluorescent staining outlining the edges of the dendrites corresponds to the VAMP-2-positive bulb-like and flattened axon terminals shown in Figure 6. (Inset) Two-fold higher magnification of the area outlined at the left. Bar = 10 μm.
Discussion
The in situ localization of molecules at the subcellular level by immuno-EM has proved to be an important method in basic and applied cell biology. In light of the increasing demand for localization information on newly discovered proteins, advances in technology and improvement of existing methods are highly desirable. Previously, we reported that the retrieval of cryosections with a mixture of sucrose/methylcellulose results in superb ultrastructural detail, which is especially of value for lipid-rich regions in the cell, such as the endoplasmic reticulum to the Golgi intermediate compartment (Liou et al. 1996; Mártinez–Menárguez et al. 1999). More recently, our colleagues Griffith and Posthuma (2002) found a method to store cryosections for an indefinite time before use, which greatly facilitates experimental planning and allows a more controlled experimental set-up. Here, we combined these methods with a novel flat-embedding approach to visualize and immunolabel cryosections of polarized cells in their natural state.
The technical difficulty we had to overcome was to remove cells from the substrate without disturbing their morphological integrity. We found that embedding of cells in a slab of 12% gelatin provided the necessary support to remove cells undamaged. Langanger and De Mey (1988) have previously used a fixed layer of 1% gelatin as a substrate for growing fibroblasts and prepared these for flat-embedding and cryosectioning. In our method, cells were grown under normal culture conditions, which results in excellent ultrastructure and also allows processing of cells that must be cultured under highly specific conditions.
Our method is especially useful to study highly polarized cells, such as hippocampal neurons. Scraping and pelleting of such cells to prepare them for cryosectioning destroys contacts between the cell body and neurites and hampers the study of axons and dendrites over longer distances. Another disadvantage of scraping is that neurites are difficult to collect from the dish because they are tightly attached. For cultured primary neurons, an additional complication of the standard method is that the number of cells is usually low, whereas in the conventional procedure considerable amounts of cells are needed to obtain a visible pellet that is manageable for further processing. These disadvantages are overcome with the flat-embedding technique. We developed our method with the principal goal of studying vesicular traffic through axons and dendrites. Because of the highly complex structure of neurons, it is essential for such studies to combine molecular topology with morphological information. On the other hand, molecular characterization of compartments may shed light on their identity and help to define subdomains of the neuron, as shown in Figure 8. Many EM studies in the neurobiology field are performed on Epon or Lowicryl sections, combined with the HRP-DAB staining method or immunogold labeling (e.g., Fletcher et al. 1991; van Lookeren–Campagne et al. 1992; Augenbaum et al. 1993; Mundigl et al. 1993; Steiner et al. 1994; Holland et al. 1998). Although sensitive, the HRP-DAB method is not quantitative and is of limited use when combinations of proteins must be localized in the same section, whereas labeling density and membrane preservation are less favorable in Lowicryl sections. The present flat-embedding method for cryosections therefore provides a valuable alternative for studies that involve quantitative localization at high resolution of (multiple) proteins in complex cell systems in their in situ orientation.
We also propose other applications for the flat-embedding technique. A sideways orientation of the gelatin slab on the specimen holder allows sectioning perpendicular to the plane in which cells grow. Thus, oriented sectioning of cells with apical and basolateral subdomains, such as epithelial cells, can be achieved. Flat-embedding can also be of use for non-polarized cells when cell–cell contacts or specialized structural adaptations are studied. In the case of non-differentiated PC12 cells, we observed that the plasma membranes were closely aligned along neighboring cells. Such information is lost using the standard procedure. The flat-embedding method provides a means to select a specific cell for cryosectioning. This can be helpful when a restricted subset of cells in a preparation needs to be studied, e.g., after microinjection. Last but not least, the method opens the valuable possibility of direct CLEM using the sensitive and quantitative immunogold labeling of cryosections as an EM approach. We have illustrated this by giving an example using epifluorescence as the light microscopic approach, but clearly this correlative method can be extended to other light microscopic techniques. For example, when antibodies against GFP or its derivatives are used, the procedure is also applicable to performance of CLEM using living cells expressing GFP-conjugated proteins as starting material, providing the exiting possibility of linking cell dynamics to high-resolution information.
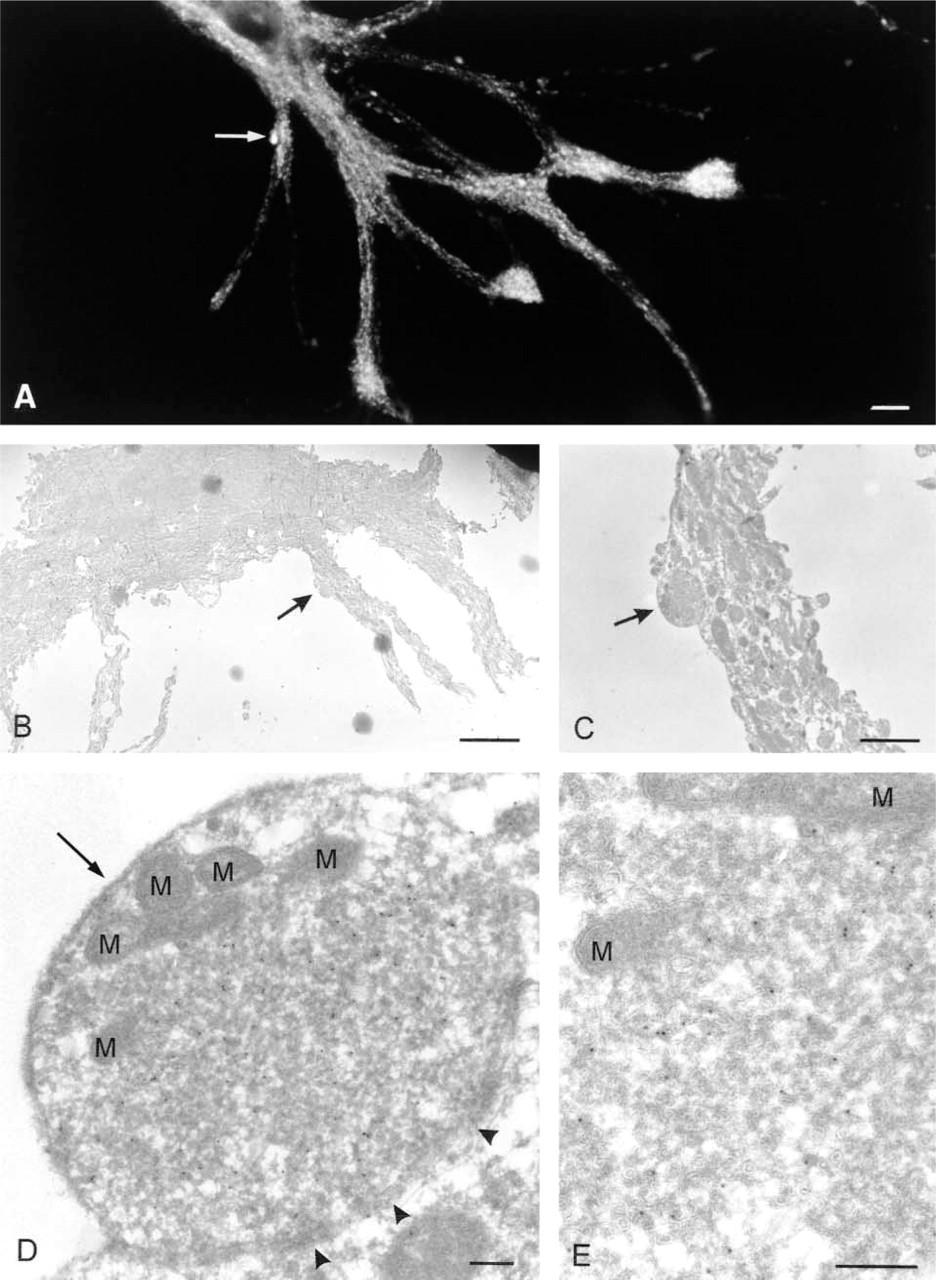
Gallery of pictures illustrating correlative light (
Footnotes
Acknowledgements
Supported by grant 805–26–183 from the Life Sciences division of the Dutch Organization of Scientific Research.
We would like to thank Rene Scriwanek and Marc van Peski for the excellent preparation of electron micrographs and for drawing Figure 1, and Dr G. Posthuma for designing the platinum wire device and assistance with ![]() . We also thank Magda Deneka and Nicolas Barois for their support with the generation of confocal micrographs and our colleagues in the Department of Cell Biology for their insightful comments.
. We also thank Magda Deneka and Nicolas Barois for their support with the generation of confocal micrographs and our colleagues in the Department of Cell Biology for their insightful comments.