
Abstract
Immunoprobes that combine a fluorescent label with a 1.4-nm gold cluster compound have been prepared by covalent conjugation with polyclonal antibody Fab' fragments. These new immunoconjugates allow the collection of two complementary sets of data, from fluorescence and electron microscopy, from a single labeling experiment. We find that incorporation of one or more fluorescein moieties into the coordinated ligands of the 1.4-nm Nanogold gold cluster label yields a stable, dual-function immunolabel in which fluorescence quenching is negligible. In a second synthetic strategy, Nanogold and fluorescein were separately covalently conjugated to Fab' fragments to yield a probe with very similar properties. With the use of Fab' fragments, the entire probe is smaller than a whole IgG molecule, and it exhibited excellent penetration properties. It was used to localize the pre-mRNA splicing factor SC35 in the HeLa cell nucleus by both fluorescence and electron microscopy.
Keywords
MICROSCOPIC STUDIES of cells and biological processes utilize a wide variety of immunoconjugates and labeled probes (Sternberger 1986; Taylor et al. 1986; Kessler 1992). An ideal label combines high resolution and easy visualization with minimal perturbation of the properties of the probe to which it is attached. A probe that combines both fluorescent and gold labels is highly desirable because it would allow unprecedented correlation between two sets of data obtained using two complementary techniques: fluorescence imaging, showing the cellular distribution of an antigen (Taylor et al. 1986; Taylor and Salmon 1989), and electron microscopy, which locates antigenic sites at the ultrastructural, macromolecular level (Handley 1989). Attempts to prepare such a probe using colloidal gold have resulted in limited success (Goodman et al. 1991). It has been found that loss of fluorescence occurs in preparations containing colloidal gold (De Brabander et al. 1985, 1986; Goodman et al. 1991). This might arise from the high extinction coefficient of the colloidal gold particles or from an electronic quenching interaction between the metallic gold and the fluorophore. Although one report describes a dual-labeled probe prepared by adsorption of fluorescein isothiocyanate-labeled protein A to 20-nm colloidal gold particles, which gave fluorescent staining similar to that found with FITC-protein A without colloidal gold (Roth et al. 1980), this does not prove that staining arises from a bifunctional probe. Colloidal gold conjugates are known to dissociate in solution (Kramarcy and Sealock 1991), and the fluorescence staining may arise from the binding of dissociated fluorescently labeled protein A. In addition, 20-nm colloidal gold conjugates may be sterically hindered from fully reaching target sites, permitting labeling with even small amounts of the much smaller dissociated fluorescently labeled protein A. It has also been found that silver-enhanced gold has weak fluorescent properties (Stierhof et al. 1992).
Gold cluster complexes used as labels offer a number of advantages over colloidal gold. Reactive groups can be incorporated during synthesis and used for covalent, site-specific attachment to biomolecules. An example is the undecagold cluster; a reactive form of this compound, with a single peripheral maleimide group, has been used to label Fab' fragments at a hinge thiol site (Hainfeld 1989). This directs the gold cluster away from the antigen-combining region; native immunoreactivity is preserved, and the resulting probe is the smallest gold immunoprobe yet to be reported.
Recently, a larger gold cluster complex, with a gold core 1.4 nm in diameter, has been developed for use in the same manner. This compound, known as Nano-gold, can be visualized in the transmission electron microscope (Hainfeld and Furuya 1992). Unlike colloidal gold, it is a neutral molecule, stable to a wide range of pH and ionic concentrations. It is stabilized by tris (aryl) phosphine ligands similar to those used in the undecagold cluster and, as with the undecagold complex, these are synthetically modified to introduce reactive groups for covalent crosslinking. This compound has been used to label IgG molecules, Fab' fragments, streptavidin, and other proteins, as well as smaller molecules, such as peptides (Segond von Banchet and Heppelman 1995; Wenzel and Baumeister 1995) and lipids, which cannot be directly labeled with colloidal gold. Monomeric conjugates are readily isolated by gel filtration, and scanning transmission electron microscopic studies confirm that aggregates are not present in these products. Goat anti-mouse Fab' fragments labeled with this compound, used as secondary probes, specifically localized MPM-2 antibodies to the centrosome and kinetochores of mitotic LLC-PK cells; in a direct comparison, a 1-nm colloidal-gold labeled antibody did not (Gilerovitch et al. 1995). Gold 1.4 nm-Fab' conjugates were found to penetrate up to 40 μm into tissue sections; the colloidal gold conjugates showed little penetration (Sun et al. 1995). In addition, the 1.4-nm gold cluster can be readily developed, using silver enhancement, in the same manner as colloidal gold conjugates (Hacker and Danscher 1994). In immunoblots, Fab' fragments labeled with this compound were found to visualize as little as 10-18 mol (1.5 × 10-13 g) of a target IgG (Hainfeld and Furuya 1992; Hainfeld and Furuya 1995). Silver-enhanced streptavidin–Nanogold with biotinylated probes in in situ hybridization was found to be more sensitive than alkaline phosphatase or peroxidase for detection of viral DNA in specimens from cervical carcinoma cases positive by solution-phase PCR for human papillomavirus (HPV) type 16 (Hacker et al. 1996). When streptavidin–Nanogold was combined with catalyzed reporter deposition (CARD), this approach gave single-copy sensitivity (Hacker et al. 1996) and was much more sensitive than conventional in situ hybridization procedures.
Synthetic modification of gold cluster coordination compounds affords unique opportunities to tailor their properties to specific applications or to introduce additional components with secondary functions. Here, we show how both a gold label and a fluorophore can be incorporated into a single probe in which fluorescence quenching is shown to be almost absent. These combined Nanogold–fluorescent probes enable simultaneous fluorescence and immunogold labeling in a single immunostaining procedure, providing a significant advantage for correlative microscopic studies.
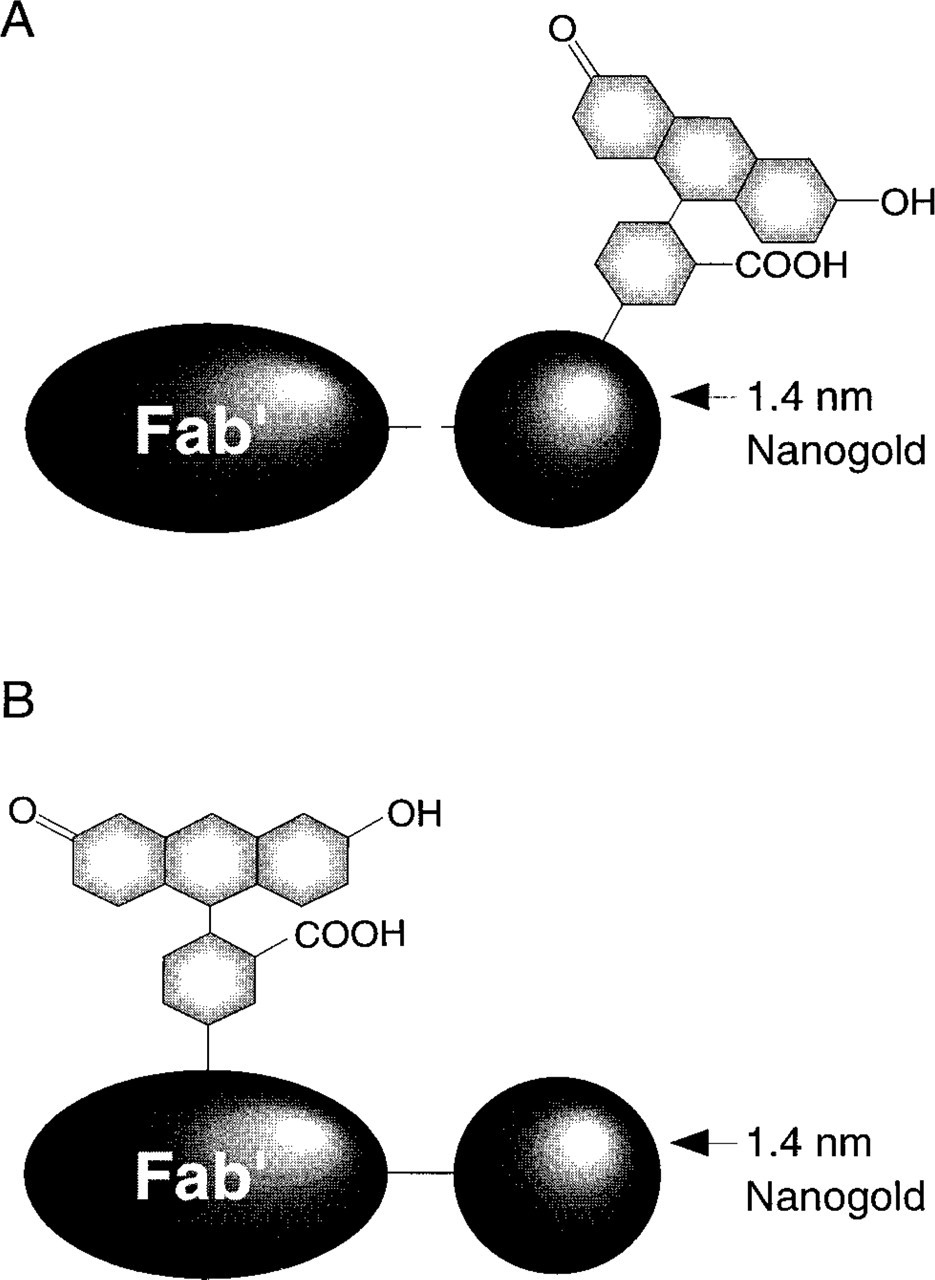
Structure of combined fluorescein and Nanogold probes. (
We describe here the preparation and properties of two types of probes (Figure 1), which combine the Nanogold cluster and fluorescent groups into a single covalently linked conjugate. In the first, fluorescein moieties were incorporated into the ligand sphere of the Nanogold cluster; this label was site-specifically conjugated to Fab' fragments (Figure 1A). In the second type, Fab' fragments were first covalently conjugated with unmodified Nanogold (i.e., without attached fluorophore), then in a second reaction this conjugate was covalently labeled with fluorescein (5- and 6-carboxy)-N-hydroxysuccinimidyl ester (Figure 1B). These conjugates were evaluated as secondary probes against a monoclonal primary antibody to localize the SC35 pre-mRNA splicing factor within the nuclei of mammalian cells. Because the fluorescein is tethered to the gold cluster in the first type, yet can be relatively remote from it in the second, it was anticipated that the nature of any interaction between the gold particle and the fluorophore could be inferred by comparing the fluorescence and spectroscopic properties of the two types of probes.
Materials and Methods
Antibodies, Biologicals, and Chemicals
Antibody Fab' fragments were prepared from F(ab')2 fragments purchased either from Jackson Immunoresearch (West Grove, PA; grades with minimal crossreactivity towards human, bovine and horse serum proteins) or Sigma (St Louis, MO). Sheep red blood cells and antibodies against them were purchased from Sigma. Triethylammonium bicarbonate 2.0 M stock was prepared by bubbling gaseous CO2 through 2.0 M triethylamine/water, followed by degasing briefly by water aspiration, then stored and diluted as necessary. Phosphine ligands were synthesized using previously described synthetic methods. GH25 and GCL90 gel filtration media were obtained from Amicon (Danvers, MA), degased using a water aspirator, and packed in glass columns (Omnifit; Cambridge UK). Pre-packed Superose-12 HR (30 cm × 1 cm) columns (Pharmacia; Uppsala, Sweden) were used for separation of labeled conjugates.
Measurements
UV/visible spectra were recorded using a Hewlett–Packard 8452A spectrophotometer, and fluorescence intensities were recorded using a Sequoia–Turner 450 fluorimeter fitted with a narrow-band 490-nm excitation and 515-nm sharp cutoff filters. HPLC was performed using a SSI model 222B or 222C titanium head pump and Pharmacia model UV-1 single-wavelength detector to monitor absorbance at 280 nm.
Preparation of a Combined Nanogold-Fluorescein Cluster Label
Fluorescein was incorporated into a combined label with Nanogold by utilizing substituents of the coordinated tris (aryl) phosphine ligands of the cluster as attachment sites for the fluorescein moieties. The ligands used for this project are shown in Figure 2. Preparations of (1) and (3) have been described previously (Hainfeld 1989) and (2) was prepared in the same manner, by reaction of 2, 3-dihydroxypropylamine with tris (p-carboxyphenyl)phosphine activated with a stoichiometric amount of 1, 1-carbonyldiimidazole in DMF. (4) was prepared by activation of (5, 6)-carboxyfluorescein with a stoichiometric amount of 1, 1-carbonyldiimidazole, followed by reaction with (1). (1) was used to introduce a primary amine for crosslinking to a protein, while ligand (2) confers water-solubility on the product. This is necessary because the hydrophobic fluoresceins greatly reduce the solubility of the cluster and otherwise might promote aggregation of the probe or nonspecific binding. The cluster was prepared using different ratios of these ligands to find the best combination of high fluorescein incorporation, solubility, and crosslinking efficiency. Best results were obtained with a compound containing (1):(2):(3):(4) in the approximate ratio 2:3:6:3.
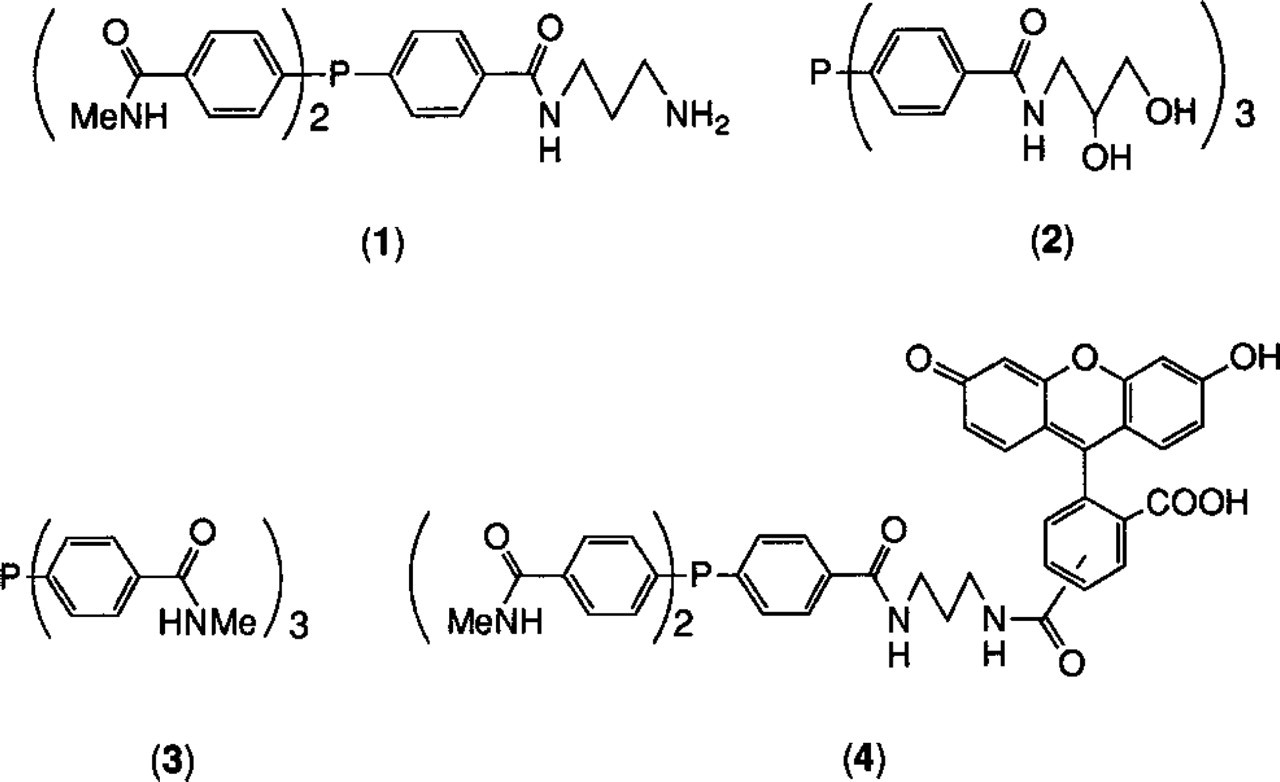
Ligands used to prepare combined fluorescein and Nanogold cluster labels.
After formation, the product was purified by HPLC on a desalting column (GH25, Amicon) eluted with 0.6 M triethylammonium bicarbonate in 20% isopropanol/water. The fluorescein-conjugated gold cluster complex was eluted first, in the void volume, followed by smaller species such as uncoordinated phosphine ligands. Fractions containing the desired product were combined, re-evaporated to dryness, then re-chromatographed in the identical manner to ensure complete removal of unattached fluorescein. Fractions containing pure cluster, identified by UV/visible absorption spectra, were combined.
Fab' Labeling with Combined Nanogold–Fluorescein Cluster Label
Goat anti-mouse F(ab')2 fragments (1.0 mg, 10 nmol) were reduced with 40 mM mercaptoethylamine hydrochloride in 0.1 M sodium phosphate buffer, pH 6.0, at room temperature (RT) for 1 hr 10 min, with 5 mM EDTA to prevent re-oxidation. The resulting Fab' fragments were separated from the reducing agent using a desalting column (GH25; Amicon).
The fluorescein/1.4-nm gold cluster was evaporated to dryness (rotary evaporator), then re-evaporated to dryness five times from methanol to remove the volatile triethylammonium bicarbonate, then converted to the maleimide as follows: 300 nmol was dissolved in DMSO (0.5 ml) and 0.1 M sodium phosphate, pH 7.50 (0.90 ml) and added to a solution of sulfo-succinimidyl 4-[N-maleimidomethyl]cyclohexane-1-carboxylate (sulfo-SMCC: Pierce, Rockford, IL; 50-fold excess) dissolved in 0.1 ml DMSO. The mixture was incubated at RT for 90 min with gentle agitation. Maleimido-cluster label was separated from excess sulfo-SMCC by HPLC on a desalting column (GH25; Amicon) eluted with 0.02 M sodium phosphate buffer, pH 6.50, with 150 mM sodium chloride and 1 mM EDTA in a mixture of 20% DMSO and deionized water. An eight- to tenfold excess of the activated cluster label was combined with the Fab' fragments. The mixture was agitated gently for 45 min at RT, then stored at 4C overnight.
Reaction products were separated by gel filtration (Superose-12; Pharmacia) to separate monomeric, labeled Fab' fragments both from larger aggregates and from unattached gold cluster labels and smaller molecules. Of the recovered Fab', at least 80% was in the form of monomeric 1:1 conjugates. Fractions containing predominantly this species were combined, reduced to minimal volume (Centricon-30 micro-concentrator; Amicon), and separated again using a second gel filtration column (GCL90; Amicon). The eluent used for both columns was 0.02 M sodium phosphate buffer, pH 7.4, with 150 mM NaCl. The fractions that comprised the first two thirds of the conjugate peak from the second separation were combined.
Conjugates were stored, after filtration through a 0.22 μm membrane filter, at a concentration of 0.08 mg/ml in 0.02 M sodium phosphate buffer, pH 7.4, with 150 mM NaCl. For storage longer than 2 weeks, 0.1% bovine serum albumin and 0.05% sodium azide were added to prevent adsorption of the Fab' to the storage vessel and to inhibit bacterial growth.
Fab' Labeling with Nanogold and Fluorescein Attached Separately
Fab' fragments were prepared as described previously. A tenfold excess of monomaleimido–Nanogold was used to label Fab' fragments; then the mixture was incubated at 4C overnight. Nanogold–Fab' was separated by gel filtration HPLC (single pass: Superose-12 column eluted with 0.02 M sodium phosphate with 150 mM NaCl, pH 7.4).
Fractions containing monomeric Nanogold–Fab' conjugates were combined, then exchanged into 0.02 M sodium phosphate with 150 mM sodium chloride, pH 8.0, and concentrated to about 0.5 ml by membrane centrifugation (centricon-30 microconcentrators, Amicon: final yield 0.5 mg labeled Fab' fragments). This solution was then combined with 5-(and 6-)carboxyfluorescein NHS ester (Molecular Probes, Eugene, OR: 0.5 mg, 100-fold excess) in DMSO (0.05 ml) and allowed to react at RT for 1 hr. The product was isolated by gel filtration (Superose-12; Pharmacia) as described above.
Conjugates were stored in 0.02 M sodium phosphate with 150 mM sodium chloride, with 0.1% bovine serum albumin (fraction V, initial fractionation by heat shock; Sigma) to prevent adhesion of the antibody to interior surfaces of the storage vial and 0.05% sodium azide to prevent bacterial growth.
Fluorescence Measurements
Fluorescence measurements in solution were made using a Turner model 415 fluorimeter equipped with a 490-nm narrow band excitation filter and 515-nm sharp cutoff emission filter. Fluorescence intensity was compared with electronic absorbance at 500 nm (the peak of the fluorescein signal from UV/visible spectroscopy). This was corrected for the absorbance of the Nanogold cluster at this wavelength by calculating the absorbance at 500 nm due to Nanogold (using its ratio to the absorbance at 420 nm where fluorescein does not absorb) and subtracting it from the absorbance value. The ratio of fluorescence:absorbance for the combined fluorescein and Nanogold conjugates was compared with that for a commercial fluorescein-labeled polyclonal F(ab')2 conjugate [goat anti-rabbit; Jackson Immunoresearch: labeled using fluorescein (5, 6) N-hydroxysuccinimide ester].
Labeling of the SC35 Pre-mRNA Splicing Factor in HeLa Cells and Microscopy
HeLa cells were grown on coverslips for 2 days and fixed, washed, and permeabilized as previously described (Spector et al. 1991). Next, the cells were incubated for 1 hr with antibody to SC35 [1:800 in PBS containing 0.1% normal goat serum (NGS)], then washed in PBS containing 1% NGS, then incubated with a 1:10 dilution of fluorescein–Nano-gold-conjugated Fab' goat anti-mouse IgG (H + L) for 1 hr at RT. At this point, fluorescence pictures were obtained using a Nikon FXA microscope equipped with a × 60/1.3 NA objective. After four 10-min washes with PBS, the cells were fixed in 1% glutaraldehyde in PBS for 15 min, followed by three 10-min washes with PBS. Before silver enhancement, the buffer was changed to 0.02 M sodium citrate buffer, pH 7.0. The cells were extensively washed in this buffer (chlorides must be removed before silver enhancement). The silver enhancement procedure was then performed in the darkroom using a Thomas Duplex sodium vapor safelight.
HQ Silver (Nanoprobes; Stony Brook, NY) was used to enhance the gold probe in the cells. HQ Silver was prepared by vortexing a 1:1:1 mixture of initiator, moderator, and activator. The backs of the coverslips were dried using filter paper and 200 μl of the silver enhancement solution was applied to the cell side of the coverslip. After approximately 15 min, or when the silver changed from clear to gray, the silver was washed off the coverslip with citrate buffer. Once silver enhancement was complete, the cells were washed extensively with citrate buffer to remove any nonspecific silver deposits and to prevent any further silver enhancement.
After washes in citrate buffer, the cells were dehydrated through a graded series of ethanol. The cells were infiltrated with a 50:50 solution of ethanol and Epon–Araldite for 18 hr followed by 100% Epon–Araldite for 8 hr. The coverslips were embedded in Epon–Araldite and placed in an oven at 60C for 48 hr to polymerize. The glass coverslips were removed using hydrofluoric acid. Silver-enhanced, gold-labeled sections were cut on a Reichert Ultracut E ultramicrotome using a Diatome diamond knife. Sections were picked up on 200-mesh copper grids and counterstained with 5% uranyl acetate for 5 min and with Reynolds lead citrate (Reynolds 1963) for 1 min. Some sections were also EDTA-regressed for 30 min (Bernhard 1969). Sections were viewed with a Hitachi H-7000 transmission electron microscope operated at 75 kV.
Immunoblot Testing of Conjugates
Conjugates were tested by immunoblots against serial tenfold dilutions of mouse IgG spotted onto hydrated nitrocellulose membrane. A modification of the procedure of Moeremans (Moeremans et al. 1984) was used:
A nitrocellulose membrane was hydrated by simmering in gently boiling water for 15 min.
One-μl dilutions of mouse IgG containing sequential tenfold dilutions from 10-8 g (10 ng) to 10-17 g (10 ag) were spotted onto membrane using a capillary.
Membrane was blocked with blocking buffer for 30 min at 45C, then washed 5 min with wash buffer.
Membrane was incubated with 5 ml of a 1:200 dilution of cluster conjugate in incubation buffer for 2 hr at RT with slow agitation.
Membrane was rinsed with buffer 3 (three times for 5 min), then PBS (three times for 30 sec).
Postfixed with glutaraldehyde, 1% in PBS (10 min), then rinsed with deionized water (twice for 5 min).
Blot was rinsed with 0.05 M EDTA at pH 4.5 (2 min) to sequester free metal ions.
Labeled blot was developed with freshly mixed LI SILVER (Nanoprobes) twice for 30 min, rinsing thoroughly with deionized water after each development, then air-dried.
Buffers
PBS. Sodium phosphate 0.01 M with 150 mM sodium chloride, pH 7.4.
Wash Buffer. PBS with 0.8% w/w bovine serum albumin (fraction V by heat shock), 0.1% w/w gelatin, type B from bovine skin, approximately 60 bloom, and 2 mM sodium azide.
Blocking Buffer. Wash buffer with additional bovine serum albumin, total BSA content 4%.
Incubation Buffer. Wash buffer with 1% w/w normal goat serum.
Labeling of Sheep Red Blood Cells and Fluorescence Measurements
A simple cell-labeling assay was developed to evaluate the fluorescence of bound antibodies. Fixed sheep red blood cells suspended to 10% hematocrit in PBS buffer (as above) were diluted to 5% hematocrit with deionized water (final volume 10 μl), then mixed with 100 μl of rabbit anti-sheep red blood cell stroma (IgG fraction) (18.5 mg/ml in 0.01 M PBS, pH 7.2) and incubated with gentle shaking for 1 hr at RT. The cells were pelleted (centrifuge, 5 min at 1900 × g), the supernatant was removed, and the cells were washed twice in PBS containing 1% bovine serum albumin (PBS–BSA). After pelleting again, the cells were resuspended in 100 μl of mouse anti-rabbit IgG (H + L) (Jackson: 1.7 mg/ml in 0.01 M sodium phosphate with 0.25 M NaCl, pH 7.6) and incubated with gentle shaking at RT for 1 hr further. After pelleting, and washing twice with PBS-BSA as described above, the cells were resuspended in 50 μl of PBS-BSA. Labeling with the combined Nanogold–fluorescent probes (goat Fab' anti-mouse IgG) was conducted in glass tubes previously blocked with blocking buffer (as described above for blotting) at 40C. Into each tube was placed 10 μl of the red blood cell suspension and 25 μl of (a) the labeled experimental anti-mouse Fab' conjugate at a concentration of 80 μg/mL, or (b) a commercial fluorescein NHS ester-labeled anti-mouse IgG at a concentration of 100 μg/ml. After shaking gently for 1 hr, the cells were washed twice with PBS-BSA buffer as described above and suspended in PBS. Their fluorescence was then measured in the fluorimeter.
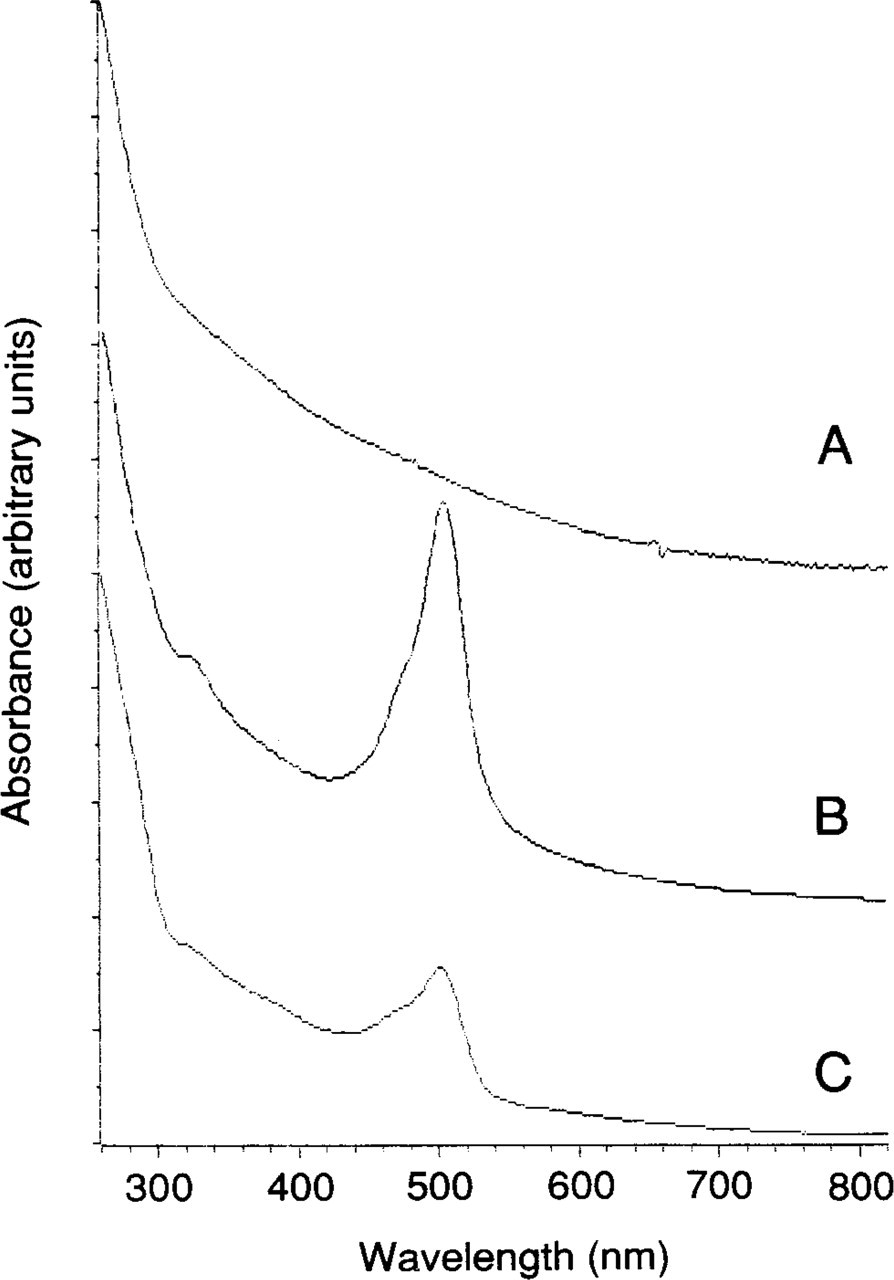
UV/visible absorption spectra of (
Results
Labeling with each of the components of the label was calculated from the UV/visible spectra of the conjugate, the UV/visible spectrum of the unattached cluster, and the spectrum of an analogous gold cluster without incorporated fluorescein. In general, the contribution of the components to the UV/visible spectrum was found to be additive. Both the unconjugated fluorescein–Nanogold cluster label and Fab' conjugates yielded UV/visible absorption spectra in which the spectral features arising from each component were clearly defined. Spectra for Nanogold (with no attached fluorophores), fluorescein-conjugated Nanogold, and a fluorescein–Nanogold–Fab' conjugate are shown in Figure 3. From comparison of the UV/visible spectrum of the fluorescein-substituted cluster with that of an identical but unsubstituted compound prepared using only nonfluorescent ligands, it was calculated that each gold cluster incorporated up to 3 fluorescein moieties but that in the labels that were conjugated to antibodies, 1–2 fluoresceins were typically incorporated per Nanogold.
From these spectra and measurements of fluorescence emission intensity, the degree to which fluorescence intensity was preserved was calculated using
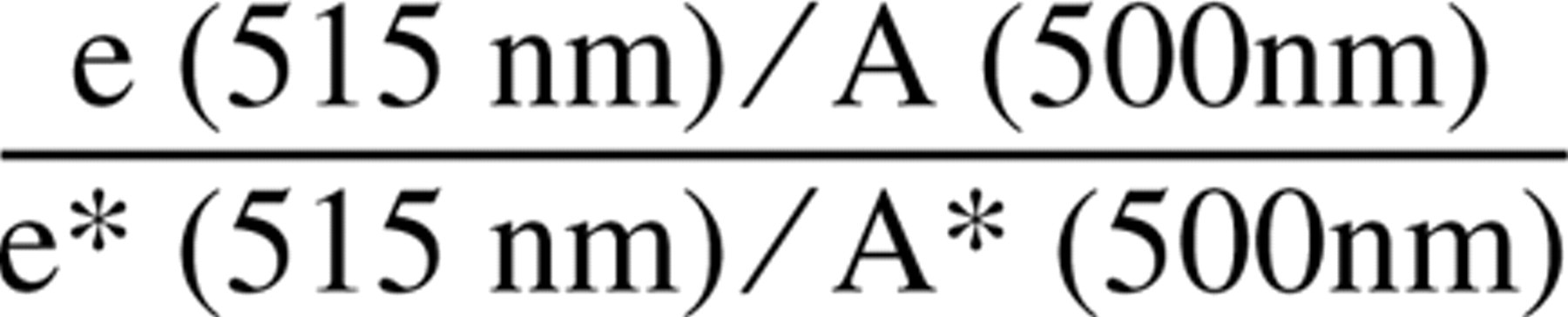
where e and A are emission and absorbance intensities for the combined Nanogold–Fluorescein–Fab' probe (A is the absorbance due to fluorescein, obtained by subtracting from the observed absorbance at 500 nm the fraction that was due to Nanogold, calculated using its known ratio to the absorbance at 420 nm where fluorescein does not absorb), and e∗ and A∗ are the corresponding values for the fluorescein-labeled F(ab')2 (containing no gold). The value found for this expression was between 0.8 and 1.0 for all conjugates examined, typically 0.83.
Figure 4A shows a fluorescence micrograph of HeLa cells in which the SC35 pre-mRNA splicing factor is labeled with the new probe as described above. Figure 5A shows an electron micrograph of a cell from the same preparation. Both are shown with controls in which the monoclonal primary antibody was absent (Figures 4B and 5B). These micrographs were obtained with probes prepared by Nanogold conjugation followed by fluorescein attachment.
Similar results were obtained from the same procedure using Fab' fragments conjugated with the combined label with the fluorescein attached directly to the Nanogold. In the electron microscope, similar low levels of background staining were found with both types of probes. Because the immunofluorescence staining required a higher concentration of the new probe than would be required for Nanogold labeling, the buffers and washing procedure were carefully optimized for the most effective removal of unbound labeled conjugates. As a result, background staining was found to be similar to that found in previous studies using nonfluorescent Nanogold conjugates.
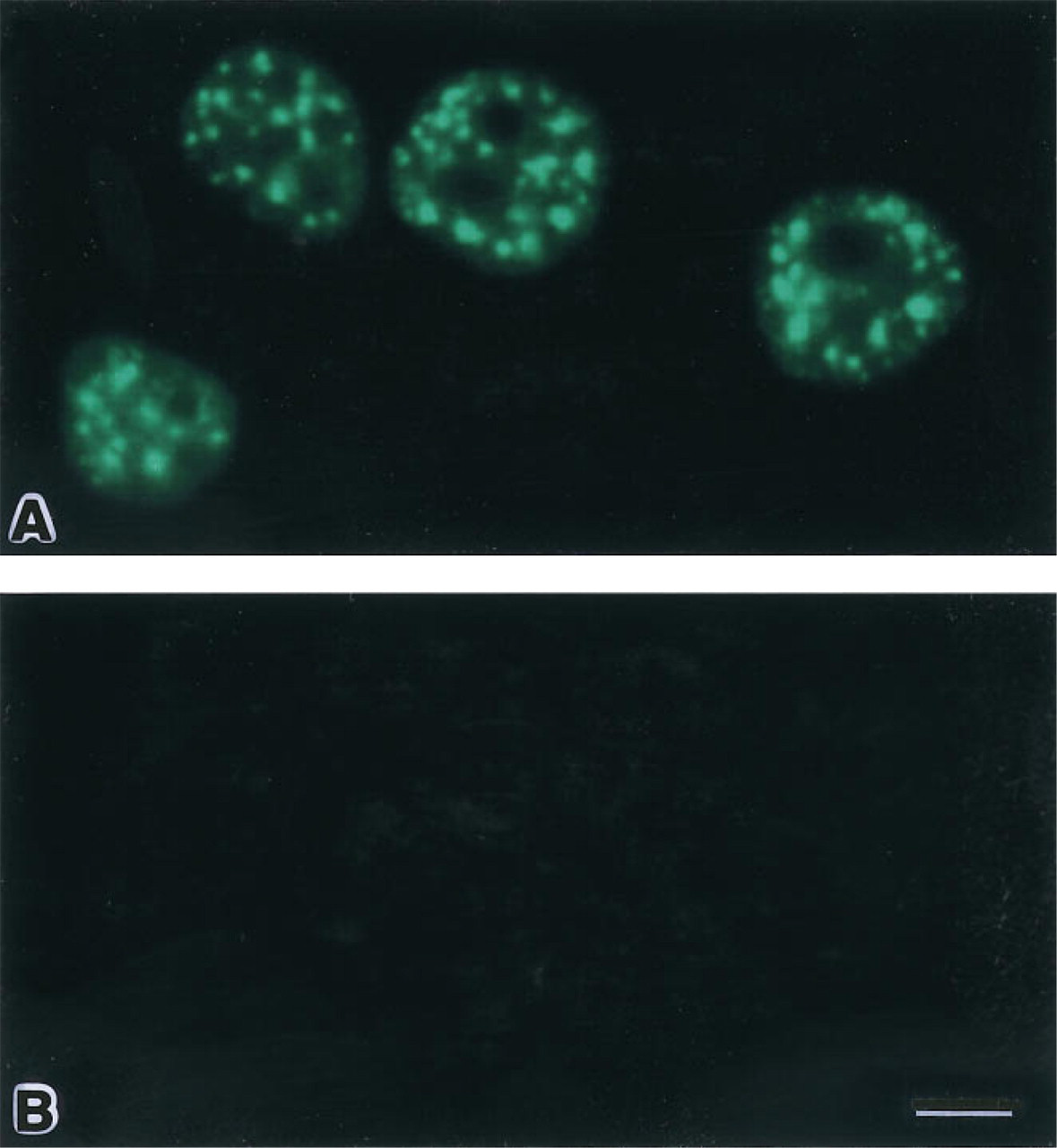
Fluorescence micrographs of labeled HeLa cells. (
The staining pattern is consistent with the distribution of SC35 found in previous studies (Fu and Maniatis 1990; Spector et al. 1991). The αSC35 antibody recognizes a 35, 000 MW protein which has been shown to be required for assembly of spliceosomes and for pre-mRNA splicing. Previously, this protein was found to localize with small nuclear ribonucleoprotein particles (snRNPs) only in the nuclear speckles and not in the surrounding nucleoplasm (Spector et al. 1991). Therefore, labeling of this factor demonstrates both the high cellular and nuclear penetration obtained with the new probe and its high signal-to-noise ratio and low background affinity. Nonfluorescent Nano-gold–Fab' probes gave similar results in electron microscopic labeling of nuclear antigens, whereas colloidal gold conjugates gave significantly lower labeling (Huang et al. 1994). Fluorescence intensities with both types of combined Nanogold–fluorescein probes were found to be about 0.4 relative to preparations labeled with a commercially available FITC-conjugated IgG secondary antibody.
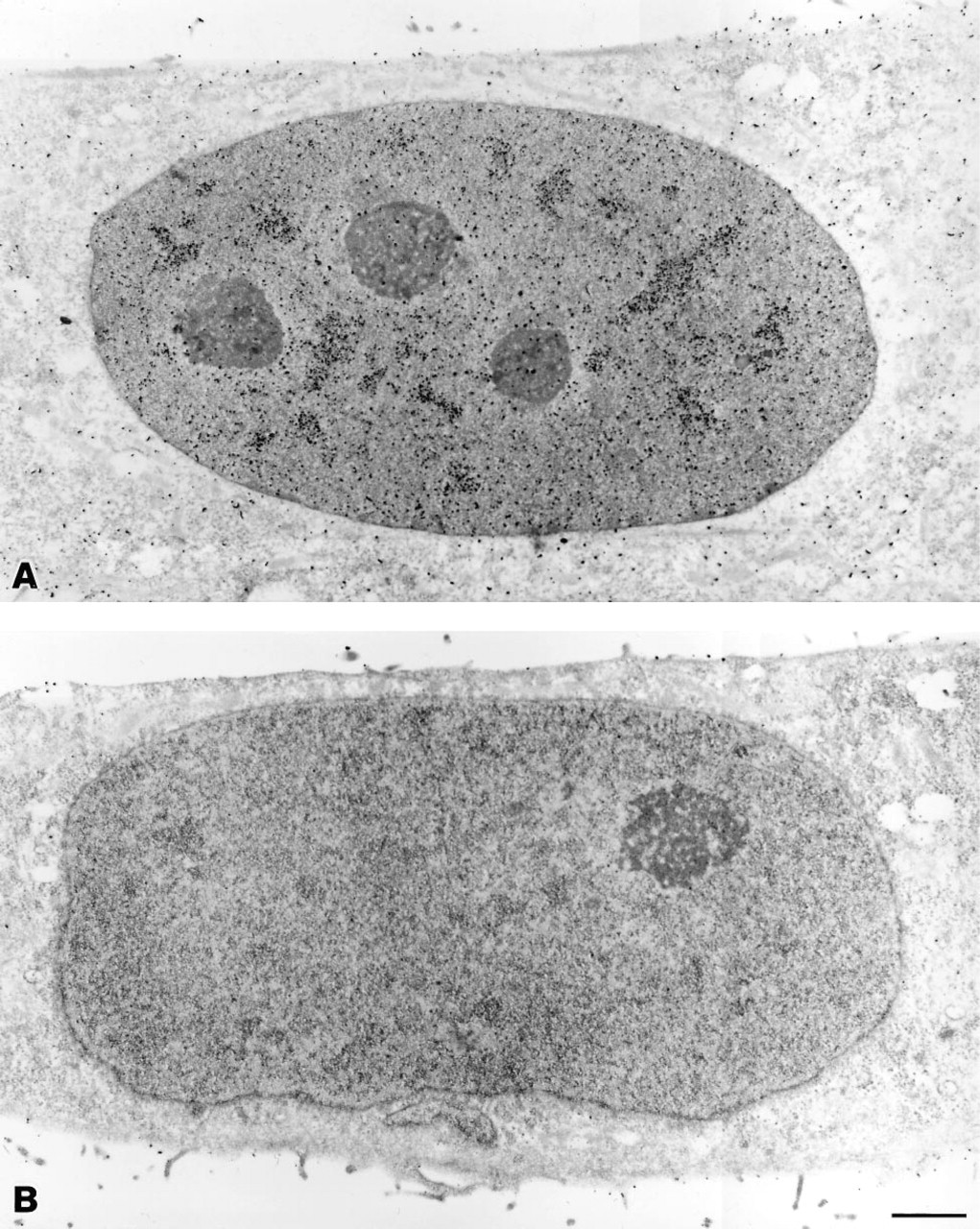
Transmission electron micrographs of labeled HeLa cells. (
Fluorescence intensities, obtained by fluorimetry, were found to be preserved for suspensions of sheep red blood cells labeled with a tertiary probe in which fluorescein was conjugated to a preformed Nanogold–Fab' conjugate as described above. Fluorescence intensities were typically close to 0.25 relative to those obtained with a commercial fluorescein-labeled IgG used as a positive control. Some of the difference might be due to the larger number of fluoresceins conjugated to the IgG compared with the experimental Fab' probe. However, probes prepared using the combined Nanogold/fluorescein label (in which the fluorescein is attached directly to the Nanogold) showed substantial reductions in fluorescence intensities. More detailed studies of their fluorescence properties are in progress to determine the exact nature of the interactions between the components of this label.
In immunoblots, conjugates prepared using both approaches successfully visualized 10 pg of rabbit IgG after silver enhancement; both the sensitivity and the level of background staining were similar to those found with nonfluorescent Nanogold in the same blot test, indicating that the gold particle is conjugated to the antibody and that antibody targeting is preserved.
Discussion
Colloidal gold probes are the preferred label for electron microscopy (Handley 1989). They are highly electron-dense, are available in a wide range of convenient sizes from 1 nm to 30 nm, and can be adsorbed to a wide range of antibodies and other targeted proteins. However, conjugation of colloidal gold is non-covalent, proceeding through the general avidity of colloidal gold particles for proteins. To prepare a stable probe with a 1:1 ratio of antibody to gold, other proteins, such as bovine serum albumin, or stabilizing agents, such as polyethylene glycol or polyvinylpyrrolidone, must be added to coat the exposed surface of the gold particle (De Mey 1983). Aggregation is also sometimes observed (Hainfeld 1990). The resulting probe is often too large to reach secluded antigens in the manner in which fluorescently labeled probes do. In addition, significant dissociation of the gold particle from the probe has been reported, even in freshly acquired preparations (Kramarcy and Sealock 1991). The use of covalently linked gold cluster conjugates has addressed these problems (Hainfeld 1989; Hainfeld and Furuya 1992), and for the first time enables the incorporation of both a gold label and a fluorophore into a single probe in which fluorescence quenching is shown to be negligible. The combined Nanogold–fluorescein probes enable simultaneous fluorescence and immunogold labeling in a single immunostaining procedure. This results in a shorter procedure, with fewer opportunities for perturbation of the specimen. Nanogold labeling can be monitored by fluorescence microscopy, thus screening samples before performance of the more extensive protocols required for EM. Labeling data from the same specimen can be more closely correlated than would be possible if two separate immunolabeling procedures were required.
An alternative method for fluorescence and electron microscope labeling with a single probe has been reported, based on the photo-oxidation of fluorescent dyes (Deerinck et al. 1994). In this novel approach, tetrabromofluorescein (eosin Y) conjugates were used to fluorescently label the sites of interest. The fluorophore was then photo-oxidized in the presence of diaminobenzidine to generate an osmophilic polymer, which was osmicated and visualized in the electron microscope. This method was used to localize poly(A)+ RNA and to visualize microtubules. In contrast to the method which we describe, this approach does not permit post-staining of cell sections, and therefore less detail of cell structure may be observed. It is also restricted to fluorophores that effectively convert the diaminobenzidine substrate, whereas combined Nanogold–fluorescent probes can be prepared using any fluorophore that can be covalently linked to the antibody–gold probe. By comparison, Nanogold produces a more punctate labeling pattern than eosin photo-oxidation, and may have advantages in quantitation and resolution of binding sites by electron microscopy. Furthermore, the gold labeling method also allows visualization of the underlying cell structure.
The stepwise probe synthesis, with chromatographic separation of the desired product after each step, confirms the attachment configuration of the probe components. With the fluorescein–Nanogold label, because only fluorophores conjugated to the gold particle are used for labeling the antibody and unattached fluorescent groups are removed chromatographically before antibody labeling, the fluorescence of the antibody conjugate must arise from the dual label, and its properties can be directly compared both with fluorescently labeled conjugates and with dual-labeled probes in which the components are separately attached. When a preformed Nanogold–Fab' conjugate is treated with amine-reactive fluorophores, these can react only with the antibody, and this provides a control for the properties of probes prepared using the combined label. The covalent crosslinking rationale enables the molecular definition to be preserved, and it is feasible, using different crosslinking reagents, to use this approach to label a wide variety of proteins and other targeted biomolecules in a site-specific manner.
The combined Nanogold–fluorescein probes behaved in a similar manner to separate fluorescent and immunogold probes. Labeling was imaged by widefield fluorescence and electron microscopy using the same procedures used for conventional fluorescent probes and 1.4-nm Nanogold-labeled probes. Neither component of the label is significantly compromised by the other. Fluorescence intensity measurements, which yielded intensities of approximately 0.4 relative to comparable commercially available fluorescein-labeled IgG conjugates, are consistent with negligible fluorescence quenching by the gold cluster. The higher fluorescence intensity values obtained with IgG likely arise because the IgG conjugate contains more fluorescein groups (3 to 4 per IgG) than the fluorescein–Nanogold–Fab' conjugate (1 to 2).
Features labeled with nonfluorescently conjugated Nanogold probes are not directly visible by light microscopy or on blots, although they can be visualized for both by silver enhancement. However, the degree of silver enhancement required is greater for light microscopy than for electron microscopy, and once developed for light microscopic observation, such samples are generally overdeveloped for electron microscope use.
In studies of luminescent molecules near metal surfaces (Chance et al. 1978; Pineda and Ronis 1985) and near small metal particles (Ruppin 1982), it has been found both theoretically and experimentally that energy transfer from the excited state of the fluorescent moiety to the surface plasmons of the metal acts to reduce excited state lifetimes, and hence steady-state intensity, when the distance between them is small (i.e., much less than the wavelength of the exciting light). Over the very small distances between the fluorescein moiety and the gold cluster in the new probes, this effect might be expected to reduce the fluorescence intensity. Although detailed fluorescence lifetime and intensity data have not yet been obtained, this effect appears to be negligible in the new probes used in microscopic localizations. This might be due to the electronic structure of Nanogold, which is believed to lack a conduction band of the type found in bulk metallic structures. Quenching, if it is to occur, must proceed through the interactions of molecular orbitals, which characteristically extend over much shorter distances (Resch and Fox 1991; Wiczk et al. 1993). However, reduced fluorescence was found in fluorimetric measurements of sheep red blood cells labeled with probes containing the combined Nanogold–fluorescein label (in which the fluorescein was directly attached to the Nanogold). It is possible that this was due to the more intense incident light excitation used in the fluorimeter. Because this probe did not exhibit superior performance in any other experiments, the second type of combined fluorescein and Nanogold probe, in which the Nanogold and fluorescein components are separately attached to the antibody, appears best suited for all the applications that we have investigated, and is now available commercially (Nanoprobes).
The described probes provide a significant advantage to correlative fluorescent and electron microscopic studies. This approach can be extended to other fluorescent moieties, which can be incorporated into gold cluster labels using similar approaches, and a number of such studies are now in progress. In the electron microscope, the characteristics of the combined probe were found to be indistinguishable from those of non-fluorescent Nanogold-labeled Fab' fragments. Penetration was the same, and silver enhancement rates and the distribution of silver-enhanced particles were essentially identical. Because the immunofluorescence staining required a higher concentration of the new probe than would be required for Nanogold labeling, the buffers and washing procedure were carefully optimized for the most effective removal of unbound labeled conjugates. As a result, background staining was found to be similar to that with nonfluorescent Nanogold conjugates.
In summary, we describe a novel reagent that effectively labels cellular antigens for both fluorescence and electron microscopy in a single immunostaining procedure. This enables direct correlative investigations at the cellular and ultrastructural levels before the more extensive processing for electron microscopy is performed on the same sample. The probe preparation methods allow the use of many commonly used fluorescent labels, and the new reagents should be useful for a variety of applications in biological microscopy.
Footnotes
Acknowledgements
Supported by a grant from the National Institute of General Medical Sciences (no. 2R44 GM48328–02) and by a grant from the Center for Biotechnology, State University of New York at Stony Brook.
We thank Dr James F. Marecek for helpful discussions.