
Abstract
Confocal laser scanning microscopy (CLSM) offers the advantage of quasi-theoretical resolution due to absence of interference with out-of-focus light. Prerequisites include minimal tissue autofluorescence, either intrinsic or induced by fixation and tissue processing, and minimal background fluorescence due to nonspecific binding of the fluorescent label. To eliminate or reduce autofluorescence, three different reagents, ammonia-ethanol, sodium borohydride, and Sudan Black B were tested on paraffin sections of archival formaldehyde-fixed tissue. Paraffin sections of biopsy specimens of human bone marrow, myocardium, and of bovine cartilage were compared by CLSM at 488-nm, 568-nm and 647-nm wavelengths with bone marrow frozen sections fixed either with formaldehyde or with glutaraldehyde. Autofluorescence of untreated sections related to both the specific type of tissue and to the tissue processing technique, including fixation. The reagents' effects also depended on the type of tissue and technique of tissue processing, including fixation, and so did the efficiency of the reagents tested. Therefore, no general recipe for the control of autofluorescence could be delineated. Ammonia-ethanol proved most efficient in archival bone marrow sections. Sudan Black B performed best on myocardium, and the combination of all three reagents proved most efficient on paraffin sections of cartilage and on frozen sections fixed in formaldehyde or glutaraldehyde. Sodium borohydride was required for the reduction of unwanted fluorescence in glutaraldehyde-fixed tissue. In formaldehyde-fixed tissue, however, sodium borohydride induced brilliant autofluorescence in erythrocytes that otherwise remained inconspicuous. Ammonia-ethanol is believed to reduce autofluorescence by improving the extraction of fluorescent molecules and by inactivating pH-sensitive fluorochromes. The efficiency of borohydride is related to its capacity of reducing aldehyde and keto-groups, thus changing the fluorescence of tissue constituents and especially of glutaraldehyde-derived condensates. Sudan Black B is suggested to mask fluorescent tissue components.
Keywords
C
Autofluorescent properties of specific tissue constituents may be of diagnostic value (Del Castillo et al. 1989; Noonberg et al. 1992). This applies, e.g., for lipofuscin granules and collagen fibers (Edwin and Jackman 1981; Verbunt et al. 1992; Belichenko et al. 1996; Banerjee et al. 1999). On the other hand, autofluorescence, either intrinsic or induced by fixation media and tissue processsing may either mask specific fluorescent signals or be mistaken for fluorescent labels (Del Castillo et al. 1989; Noonberg et al. 1992).
Various histochemical techniques for the removal of autofluorescence have evolved. Ammonia-ethanol has been applied to remove some of the formaldehydederived artifacts (Kardasewitsch 1925), sodium borohydride (borohydride) was used to control autofluorescence of glutaraldehyde-related condensates (Johnson 1987: Baschong et al. 1997, 1999; Southern et al. 2000), and various dyes, including Sudan Black B (sudan), have been used to block autofluorescence in nerve tissue (Romijn et al. 1999). However, a generally applicable recipe is still missing, particularly for routine paraffin sections of formaldehyde-fixed tissue.
Here we assessed the effects of the three reagents, ammonia-ethanol, borohydride, and sudan on autofluorescence in paraffin sections of archival formaldehyde-fixed and decalcified human bone marrow before fluorescence labeling. We compared them with the effects on archival myocardium and cartilage and with frozen sections of the same tissue types. The aim of our study was to minimize autofluorescence to enable high-resolution CLSM also in formaldehydefixed archival tissue and to elucidate the influence of tissue type, fixation medium, and tissue processing.
Materials and Methods
Unless otherwise specified, all reagents were of analytical grade and purchased from Sigma (St Louis MO), Fluka (Buchs, Switzerland), or Merck (Darmstadt, Germany), and all steps of the procedures were done at ambient temperature (20–25C).
Tissue Sections
Paraffin blocks, stored for 6 months to 3 years, were retrieved from our archival files of human bone marrow and myocardial biopsies fixed in neutral buffered formaldehyde (4% final concentration) for 12–24 hr, cut to 8-μm-thick sections, and mounted on sialinized slides. After fixation, bone marrow core biopsies were decalcified overnight in 5% trichloroacetic acid under continuous agitation. Cartilage specimens, a gift from J. Seidel (MIT; Cambridge MA), were derived from articular bovine chondrocytes grown on a scaffold of polyglycolic acid (Freed et al. 1998), fixed in formaldehyde (10% final concentration) for 2 hr, and processed for paraffin sections. For removal of paraffin, the slides were immersed in xylene (twice for 20 min), followed by graded ethanol (15 min in 100%, twice for 15 min in 96%, 5 min in 80%, 10 min in 70%, 10 min in 50% ethanol) and transferred to modified Hanks' buffer [MHB: Hanks' buffer without calcium but with 2 mM EGTA and 5 mM MES (2-morpholino-ethanesulfonic acid), pH 6.2–6.4, supplemented with 0.005% NaN3 (Small and Celis 1978)]. Unless immediately processed, the slides were stored for up to 2 weeks in MHB at 4C. Before labeling, the sections were permeabilized for 30 min in MHB containing 2% octyl-POE (n-octylpolyethoxyethylene; Alexis, San Diego, CA) and retransferred to MHB.
Frozen sections of postmortem human bone marrow taken from the femoral medulla required no decalcification. They were cut to 8-μm thickness, postfixed for 1 hr either in formaldehyde (4% final concentration) or in 2.5% glutaraldehyde, and air-dried until further processing.
Sections were treated with ammonia-ethanol, borohydride, or sudan and by combinations thereof as follows.
Ammonia-Ethanol
While rehydrating the deparaffinized sections in graded alcohol, the slides were immersed for 1 hr in 70% ethanol supplemented with 0.25% NH3, and rehydration was resumed by immersion in 50% ethanol for 10 min, after which the sections were transferred to MHB.
Borohydride
Deparaffinized sections were immersed in ice-cooled freshly prepared MHB supplemented with 10 mg/ml borohydride for 40 min, washed three or four times in MHB, and stored at 4C in MHB until further processing.
Frozen sections were treated accordingly after permeabilization as described above.
Sudan
After fluorescence labeling, the slides were immersed for 30 min in 70% ethanol supplemented with 0.1% Sudan Black B (Merck). The removal of excess sudan from the slides proved decisive. Sudan had to be wiped off manually from the back and along the edges of the slide with a soft paper, and the front required a jet wash with MHB. Subsequently, slides were immersed for 10 min in MHB and mounted. Omitting this procedure resulted in sudan precipitates appearing as subcellular black grains in the differential interference contrast (DIC) image.
Postfixed frozen sections were treated accordingly for autofluorescence control, observing the sequential order of borohydride, ammonia-ethanol, and sudan.
Immunofluorescence Labeling
The slides stored in MHB were conditioned for 20 min with 50–75 μl of MHB containing 10% swine serum (Jackson Immune Research Laboratories; West Grove, PA) and incubated for 1 hr with the primary antibody diluted in MHB. The sections were washed three or four times for 5 min with aliquots of 100–200 μl of MHB, incubated for 1 hr with secondary antibody diluted in 75 μl MHB, and washed as above. Primary antibodies used were directed against β-tubulin (clone KMX-1, 1:50; Boehringer, Mannheim, Germany) and the secondary antibody was donkey anti-mouse labeled by Cy2 (1:400, 488 nm; Amersham, Poole, UK).
All sections were mounted with Mowiol-1188 (Hoechst; Frankfurt, Germany) supplemented with 0.75% of the antifading agent n-propyl-gallate (Baschong et al. 1999), protected with a coverslip, dried overnight, and stored in the dark until viewing. The effects of treatment for autofluorescence control were assessed by a Leica TCS 4-D CLSM. Fluorescence image stacks were registered as 0.3–0.5-μm optical sections in parallel in the 488-nm (green), 568-nm (red), and 647-nm (blue) channels. The series of DIC images were registered in a subsequent scan. Fluorescence of treated sections was compared to that of untreated sections cut from the same tissue block and for each type of tissue and fixation medium, respectively.
Results
Bone Marrow Paraffin Sections
In untreated paraffin sections of formaldehyde-fixed, decalcified bone marrow, trabecular bone and cytoplasmic granules of myeloid cells displayed autofluorescence (Figures 1A and 1B). Treatment with ammoniaethanol reduced bright autofluorescence of myeloid cells most efficiently regardless of the wavelength (488 nm, 568 nm, or 647 nm) used for excitation (Figure 1C). Reduction of autofluorescence was observed at 488 nm and 568 nm also after treatment with sudan (Figure 1D), but not at 647 nm, where sudan's own fluorescence became apparent (Romijn et al. 1999). A near to total absence of autofluorescence was attained by treatment with ammonia-ethanol and sudan (Figure 1E). This combination suppressed autofluorescence of trabecular bone and myeloid cells when exposed at 488-nm and 568-nm light, but not at 647 nm, where ammonia-ethanol alone performed best. To our surprise, treatment with borohydride induced bright autofluorescence in erythrocytes that had otherwise remained inconspicuous (Figure 1F). This undesirable effect was slightly diminished by adding ammonia-ethanol (Figure 1G) or sudan (Figure 1H) and even better if both reagents were combined (Figure 1I). Nevertheless, borohydride treatment once applied, autofluorescence in erythrocytes could not be completely abolished (Figure 1I).
The complex organotypical histoarchitecture of the bone marrow was more accurately appreciated in DIC, where trabecular bone, cells of various hematopoietic cell lines, and stromal structures, including adipocytes, were easily distinguishable (Figure 2A).
Bone Marrow Frozen Sections
In untreated formaldehyde-postfixed frozen sections of non-decalcified bone marrow, autofluorescent cytoplasmic granules were seen in myeloid cells (Figure 2C), similar to decalcified paraffin sections (Figure 2B). Sudan decreased unwanted autofluorescence most effectively (not shown), again when exposed at 488-nm and 568-nm wavelengths, but not at 647 nm, for which ammoniaethanol proved superior. Borohydride was inefficient in this respect (Figures 2C and 2F).
In glutaraldehyde-postfixed untreated frozen sections of non-decalcified bone marrow, myeloid cells displayed bright autofluorescence (Figure 2D). The latter was considerably reduced by borohydride (Figure 2E), and occasionally even more so when combined with ammonia-ethanol and/or sudan (not shown).
Myocardial Paraffin Sections
In untreated paraffin sections of formaldehyde-fixed myocardium, lipofuscin granules showed as autofluorescent particles, whereas the sarcoplasmic body of myocardiocytes remained dim (Figure 2G). Treatment with ammonia-ethanol and borohydride, either separately or combined, highlighted the sarcomeric Z-lines at 488 nm and 568 nm (Figure 2H). They were not perceivable at 647 nm (not shown), whereas autofluorescence of lipofuscin granules was present at all three wavelengths. Treatment with sudan, however, completely removed autofluorescence from myocardiocytes (Figure 2I).
Cartilage Paraffin Sections
In untreated paraffin sections of engineered bovine cartilage, weak autofluorescence was present in the intercellular space. It was related to the scaffold made of polyglycolic acid, which was used for cultivation. Whereas borohydride and ammonia-ethanol induced cytoplasmic fluorescence in chondrocytes, optimal control of autofluorescence was achieved by combining no less than all three reagents, i.e., ammonia-ethanol, borohydride, and sudan (not shown).
Interference with Fluorescence Labeling
To illustrate the applicability of autofluorescence control, an example of a myeloproliferative disorder was selected from our archival bone marrow files and labeled for β-tubulin immunofluorescence. In tubulinlabeled untreated paraffin sections exposed at 488 nm, an ill-defined cluster of green fluorescence was registered (Figure 3A, arrow). When the same area was exposed at 568 nm, distinct autofluorescence was displayed by cytoplasmic granules in myeloid cells (Figure 3B, arrow). The presence of these autofluorescent granules at 488 nm was camouflaged by the immunofluorescent label (Figure 3A, arrow). After treatment with ammonia-ethanol and sudan, stromal cells and myeloid cells displayed a green fluorescent cytoplasm, indicating the presence of tubulin (Figure 3C), whereas unwanted autofluorescence was absent (Figure 3D). In conjunction with proper autofluorescence control, CLSM provided a high-resolution insight into the cytoskeletal architecture of microtubules in both interphase (Figure 3C') and mitotic cells (Figure 3C”).
Discussion
Interference by autofluorescence is one of the major shortcomings of CLSM in immunofluorescence analysis. This phenomenon may either be intrinsic, due to fluorescent structures in cells and interstitial tissue, or may be induced by fixation media and tissue processing techniques. Yet another source of unwanted fluorescence relates to nonspecific binding of the fluorescent marker (Del Castillo et al. 1989). Removal of the resulting background fluorescence is commonly addressed in the protocols for immunofluorescence labeling and was not considered. Although a generally applicable recipe for the control of autofluorescence is still lacking, various methods have evolved for a limited number of tissue types and for special studies, such as analysis of the cytoskeleton in glutaraldehydefixed specimens (Baschong et al. 1997,1999).
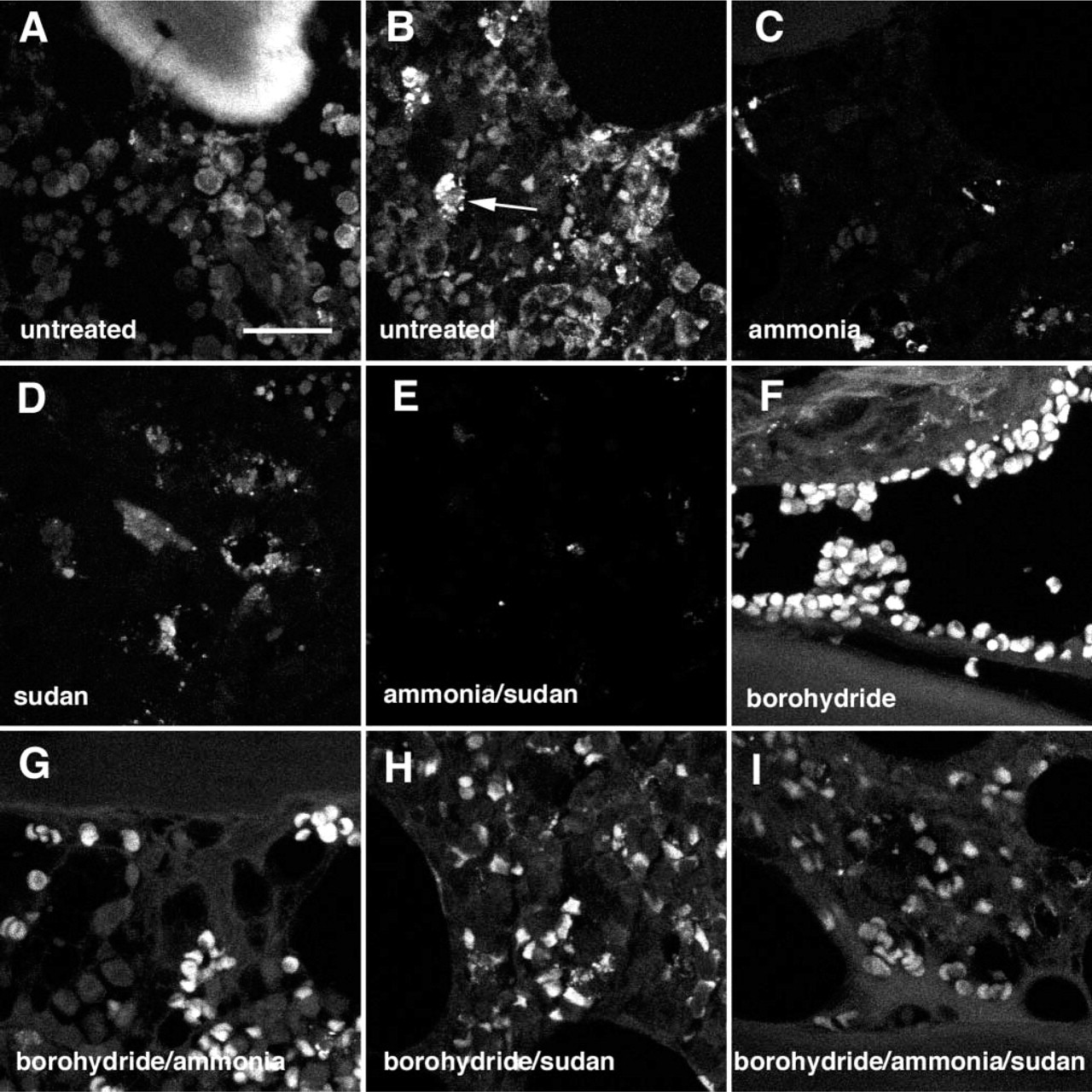
Histochemical control of autofluorescence in archival paraffin sections of formaldehyde-fixed human bone marrow. Confocal laser scanning microscopy (CLSM) of unlabeled sections, composite images of 15–30 optical planes of sections (0.3 μm) exposed at 488 nm. Untreated controls; autofluorescent trabecular bone (
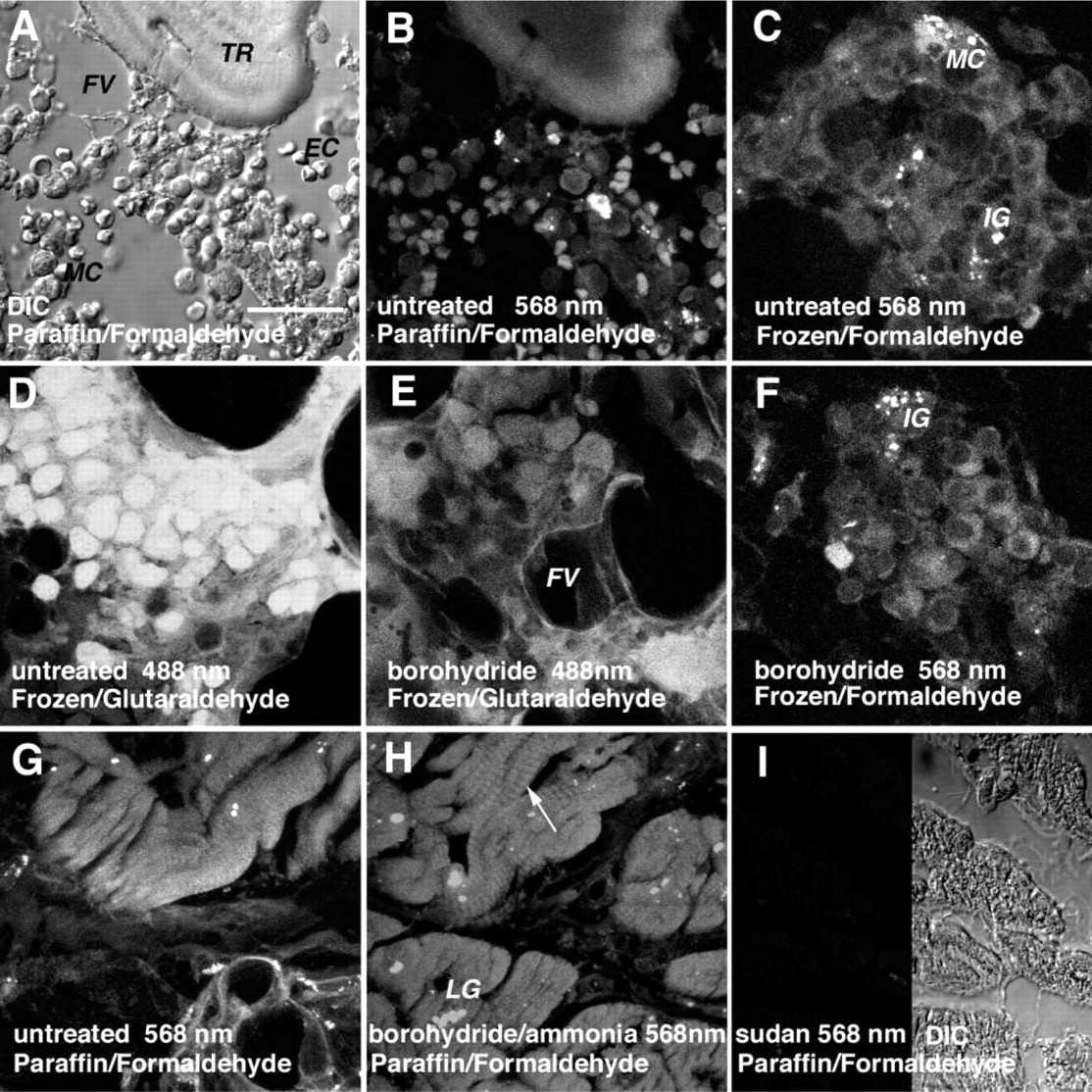
Influence of tissue type, fixation media, and processing on histochemical control of autofluorescence. (
At least three different strategies can be distinguished in the control of autofluorescence: (a) extraction of the autofluorescent constituent by dissolution; (b) chemical modification of the fluorochrome; and (c) masking of the autofluorescent structure by appropriate staining.
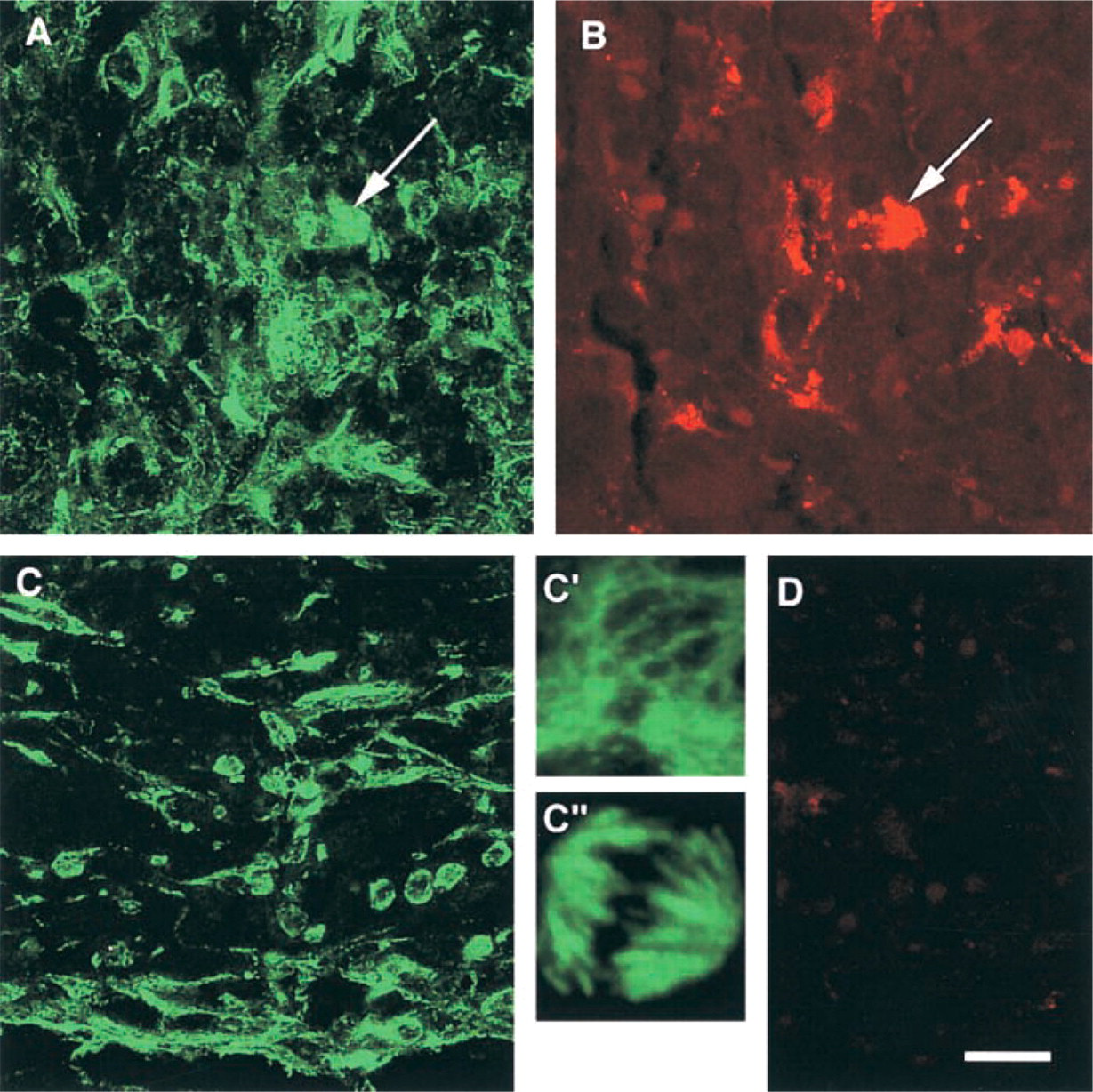
Interference of immunofluorescence labeling with specific autofluorescent structures. Archival paraffin section of formaldehyde-fixed bone marrow in an example of cellular phase of idiopathic myelofibrosis labeled with anti-β-tubulin and Cy2 (488 nm). Cluster of ill-defined green fluorescence in untreated section labeled for tubulin and exposed at 488 nm (
Ammonia-ethanol may promote the removal of precipitates formed during bone decalcification by trichloroacetic acid. It may dissolve negatively charged lipid derivatives, phenols or polypenols, and degrade weak esters by hydrolysis. Improved extraction and dissolution by ammonia-ethanol of autofluorescent entities are the putative modes of action.
Borohydride interacts with aldehydes and ketones by reducing both reactive groups to the respective alcohols. Apart from its usefulness in removing the excess of aldehyde after tissue fixation, borohydride also efficiently reduces processing-related fluorescence in glutaraldehyde-fixed tissue (Baschong et al. 1997,1999). The latter effect is believed to be the result of interaction with aldehyde and ketogroups of homo- and heterocondensates of glutaraldehyde emerging in the course of glutaraldehyde polymerization (Johnson 1987; Southern et al. 2000). However, borohydride may also affect oxidized aromatic compounds, including quinone-like structures, polyphenols, and flavonoids.
Unlike ammonia-ethanol and borohydride, sudan masks autofluorescent structures (Romijn et al. 1999). Its veiling activity on autofluorescence without interfering with the surface fluorescent label resembles the effect of counterstains such as Eriochrome Black T (Kittelberger et al. 1989) or picrosirius red (Brotchie et al. 1999) used for fluorescence control in frozen sections of blood vessels or nerves, respectively.
With respect to the difference of action among the three agents tested, there is no surprise at their variable effects depending on tissue type, fixation, processing, and wavelength of excitation light. The comparative assessment of ammonia-ethanol, borohydride, and sudan on sections cut from the same tissue blocks should allow a ranking of the agents' performances.
Ammonia-ethanol proved most efficient on paraffin sections of formaldehyde-fixed, decalcified bone marrow, but much less so on frozen sections of non-decalcified bone marrow. Its effect on autofluorescent granules of myeloid cells was obvious, but its performance against fluorescence in formaldehyde-fixed bone marrow and myocardium was far below our expectations. Introduced by Kardasewitsch in 1925, ammonia-ethanol proved nevertheless a powerful suppressor of autofluorescence in paraffin sections of formaldehyde-fixed, decalcified bone marrow. Deposits formed during decalcification by trichloroacetic acid and formaldehyde-induced condensates may be thoroughly dissolved (Kardasewitsch 1925), and degradation of pH-sensitive chromophores may further add to the efficacy of ammonia-ethanol.
Borohydride is now a confirmed requisite for autofluorescence control in glutaraldehyde-fixed tissue (Baschong et al. 1999). A paradoxical effect, however, turned up in formaldehyde-fixed tissue. Whereas borohydride contributed little to the suppression of autofluorescence in cartilage paraffin sections and in frozen sections of bone marrow and myocardium, it induced bright fluorescence in erythrocytes. A similar effect had been observed by CLSM in tissue remnants of explanted dental implants (Baschong et al. 2001). At present we cannot allocate this effect to a specific stage of red blood cell maturation. In addition, borohydride also enhanced the discernability of sarcomeric Z-lines in paraffin-embedded myocardiocytes.
Sudan was generally effective for fluorescence control in formaldehyde-postfixed frozen sections of bone marrow and in myocardial paraffin sections. Masking of brightly fluorescent lipofuscin granules by sudan is observed at a concentration as low as 0.1% in our experience, but may require a higher concentration of 1% in neural tissue (Schnell et al. 1999).
The results of the present study highlight the fact that there is no general recipe available for the control of autofluorescence. Success was found in a tactical approach that can be summarized as choice of the appropriate reagent(s) by trial and error, either single or combined, mutually designed for the specific tissue type, fixation medium, processing technique, and wavelength of excitation light.
Thus far, CLSM has been applied on routine paraffin sections for visualizing the three-dimensional architecture by staining cells with fluorescent dyes (Tekola et al. 1994; Liu et al. 1997). Here we demonstrated the versatility of CLSM in immunofluorescence labeling of a complex organ such as bone marrow in which different compartments and cell lines are interactive under normal, reactive, and neoplastic conditions, and gave proof for an insight even at subcellular resolution. Owing to proper control of autofluorescence, future analysis and reference documentation of follow-up on archival files by multiple fluorescence labeling are now a real option.
Footnotes
Acknowledgments
Supported by the Swiss Cancer League grant no. SKL-00653-2-1998/KFS 962-9-1999.
We thank Dr L. Landmann (Institute of Anatomy, University of Basel) for the complimentary use of CLSM facilities.