
Abstract
Spatial analysis of the histoarchitecture and photographic documentation at high resolution are the principal advantages of confocal laser scanning microscopy (CLSM) over conventional fluorescence microscopy (CFM) if combined with appropriate software. Restrictions for the use of CFM and CLSM, on the other hand, include nonspecific background fluorescence, fading of photolabile fluorochromes, and both tissue-specific and fixation-induced autofluorescence. Most of those shortcomings can now be avoided. Autofluorescence, the most limiting factor of high-resolution CLSM, was recently controlled also for paraffin sections of archival formaldehyde-fixed tissues. This allowed the present study on cytoskeletal fibers and extracellular matrix proteins in both neoplastic cells of myeloproliferative disorders and in medullary stromal cells using CLSM under proper autofluorescence control. By multiple fluorescence labeling, we found that the intracellular smooth muscle α-actin (SMA) fibers and the two extracellular adhesive matrix proteins tenascin and fibronectin vary in their presence in stromal and/or myeloid cells according to the degree of bone marrow fibrosis in chronic myeloproliferative disorders (CMPDs). CLSM offers further insight in our attempts to understand a complex interplay between the two cellular compartments.
Keywords
C
Materials and Methods
Paraffin blocks of bone marrow biopsy specimens fixed for 12–24 hr in neutral buffered formaldehyde (4% final concentration) and decalcified overnight at room temperature (RT) in 5% trichloroacetic acid under continuous agitation were retrieved from our archival files. For the present study, five examples of CMPD, i.e., chronic myeloid leukemia (CML) and idiopathic myelofibrosis (IMF, or myelofibrosis with myeloid metaplasia) at various stages of fibrosis (three CML with slight, one CML and one IMF with moderate to severe fibrosis) were selected, and cut to 8-μm-thick slides. Normal bone marrow served as control, and original conventional histochemical stains including hematoxylin and eosin, Giemsa, and Gomori's reticulin were reviewed for comparison. For removal of paraffin, the slides were immersed twice for 10 min in xylene, followed by graded ethanol (10 min in 100%, twice for 10 min in 96%, 10 min in 80%, 60 min in 70% containing 0.25% NH3, 10 min in 50% ethanol). Deparaffinized sections were transferred to modified Hanks' buffer [MHB: Hank's buffer without Ca but with the addition of 2 mM EGTA and 5 mM MES (2-morpholino-ethanesulfonic acid), preserved with 0.005% NaN3 and adjusted to pH 6.2–6.4] until further processing (Small and Celis 1978).
The slides removed from the MHB storage buffer were treated for 30 min at 37C with hyaluronidase (1 mg/ml testicular hyaluronidase; Sigma, St Louis, MO), conditioned for 20 min in 50–75 μl of MHB containing 10% swine serum (Jackson Immunoresearch; West Grove, PA) and immersed for 60 min in the primary antibody diluted in MHB. Sections were washed three or four times for 5 min with 100–200 μl of MHB, incubated for 60 min with the secondary antibody diluted in 75 μl MHB, and washed as above. The primary antibodies chosen for the study of bone marrow stromal cells were directed against tenascin (1:50, polyclonal rabbit anti-chicken antibody recognizing all three isoforms of the tenascin molecule; a kind gift from M. Chiquet, ME Muller Institute, Berne, Switzerland) (Chiquet 1999; and personal communication), smooth muscle α-actin (SMA) (1:500, clone 1A4; Sigma), fibronectin (1:300, polyclonal anti-rabbit antibody; Calbiochem, La Jolla, CA), myeloperoxidase (MPO, 1:1000, polyclonal; DAKO, Glostrup, Denmark) and CD68 (1:50, clone PG-M1; DAKO). Secondary antibodies were Alexa 488-nm goat anti-mouse or Alexa 488-nm goat anti-rabbit (both 1:800, 488-nm; Molecular Probes, Eugene, OR) and Cy3 donkey anti-mouse or Cy3 goat anti-rabbit (both 1:3000, 568-nm; Jackson Immunoresearch). After fluorescence labeling, the slides were immersed for 30 min in 70% ethanol supplemented with 0.1% Sudan Black B (Merck; Darmstadt, Gremany), washed with MHB and transferred into MHB. All sections were mounted with Mowiol-1188 (Hoechst; Frankfurt, Germany) containing 0.75% of the anti-fading agent N-propyl-gallate (Baschong et al. 1999), dried overnight, and stored in the dark until microscopic analysis.
For removal of autofluorescence, immersion for 60 min in 70% ethanol supplemented with 0.25% NH3 while rehydrating deparaffinized sections in graded ethanol (see above) proved optimal if combined with Sudan Black B on fluorescence labeling (Baschong et al. 2001). After this step, the rehydration procedure was resumed by immersion in 50% ethanol for 10 min and transfer to MHB. The removal of excess Sudan Black from the slides proved to be decisive. It had to be wiped off manually from the back and along the edges of the slides with a soft paper, while the front required a jet wash with MHB (Baschong et al. 2001). This procedure was followed by the transfer of the slides into MHB for 10 min before mounting.
The sections were viewed in a Leica TCS 4-D CLSM. Fluorescence image stacks were registered as 0.3–0.5-μm optical sections in parallel in the 488-nm (green) and 568-nm (red) channels. The series of differential interference contrast (DIC) was registered in a subsequent scan (Baschong et al. 2001).
Results
In normal human bone marrow, the delicate network of stromal cells was inconspicuous when examined for the presence of the extracellular matrix proteins tenascin and fibronectin, and SMA was recognized only in vascular smooth muscle cells. In CMPD, however, tenascin was found not only in delicate interstitial deposits surrounding stromal cells particularly in paratrabecular areas, but also in the cytoplasm of neoplastic myeloid cells at a stage of insignificant fibrosis (Figures 1A-1D upper halves, and Figure 1E). With increasing interstitial fibrosis, tenascin appeared in a dense network, most prominent around trabecular bone (Figure 1, lower half). In CML, severe interstitial fibrosis was an occasional finding at late stages, and was characterized by tightly woven tenascin deposits (Figure 2). In normal bone marrow, SMA was confined to smooth muscle cells of medullary vessels and was not found in myeloid cells. In myeloproliferative disorders, however, SMA became evident in paratrabecular areas but was also found in a few medullary stromal cells. This delicate pattern of distribution was observed in only a single example of CML at a stage of slight stromal fibrosis (Figure 1E). In contrast, a loose but more prominent network of stromal SMA became evident with increasing medullary fibrosis (Figure 2C). Advanced medullary fibrosis in CML was also associated with an increase of stromal cells co-expressing SMA and tenascin, although this pertained only to a minority of stromal cells (Figure 2D).
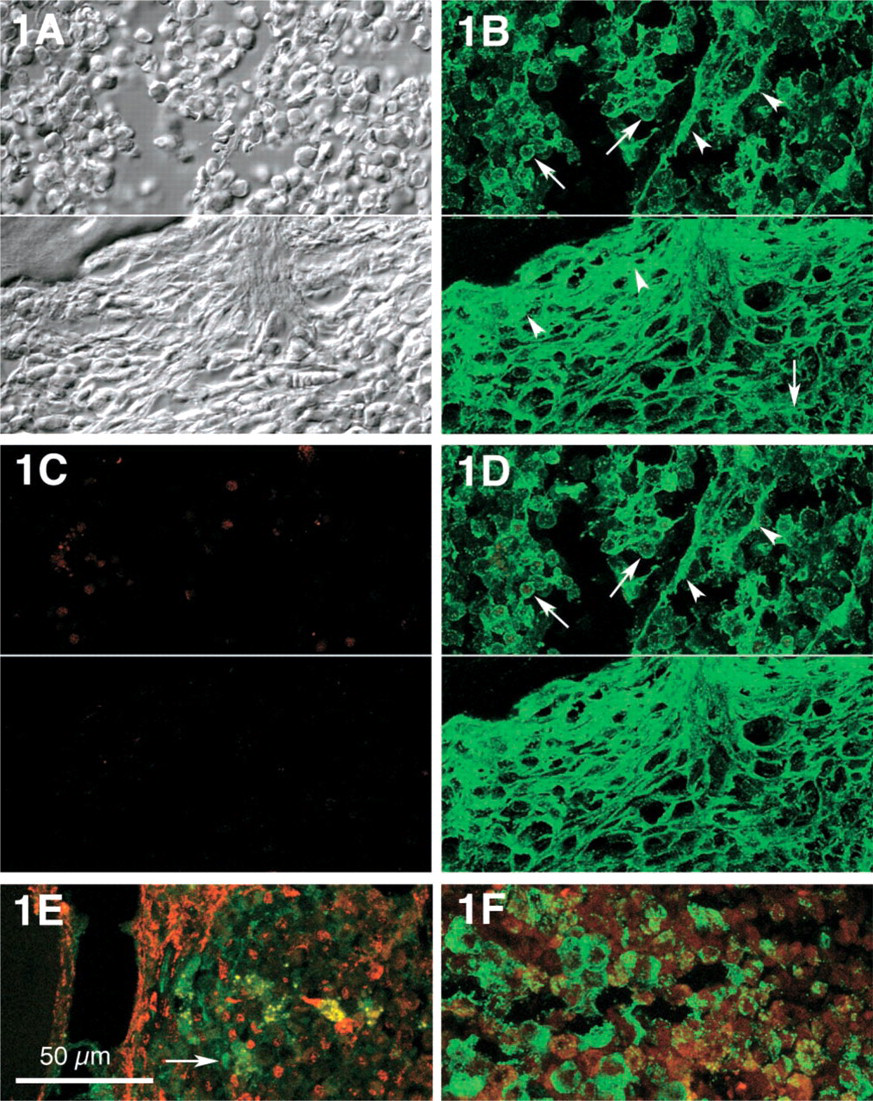
(
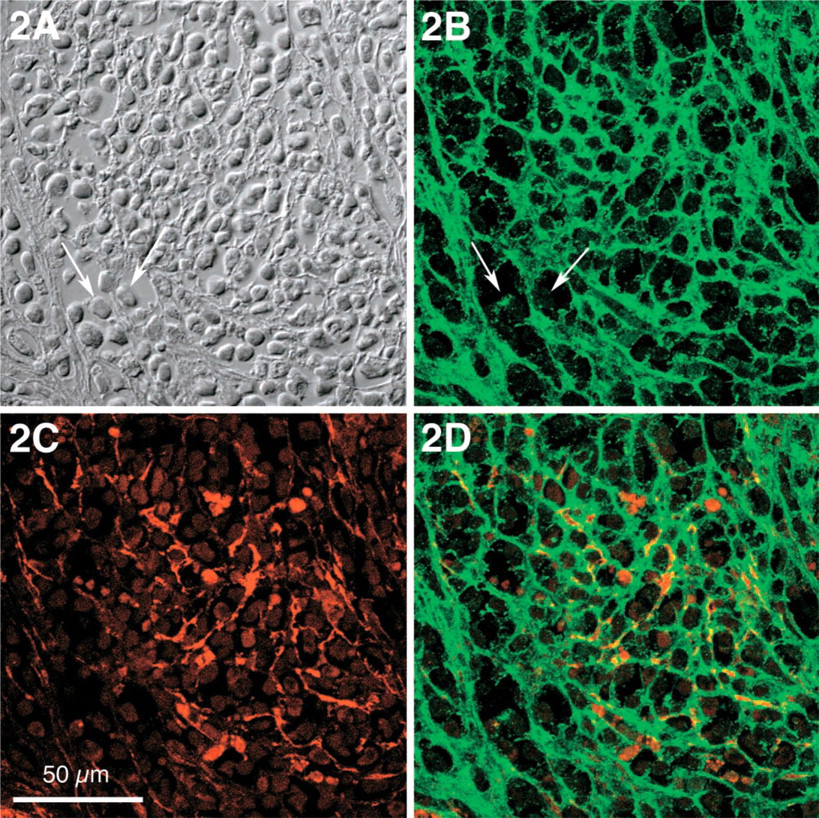
CML, centromedullary area with severe stromal fibrosis. (
In IMF, coarse deposits of SMA were predominant in stromal cells, while paratrabecular SMA appeared rather delicate but in condensed deposits (Figure 3B). Fibronectin in IMF, unlike tenascin, displayed a purely interstitial stromal pattern and was seen spanning parallel to osseous trabecules (Figure 3C) as well as in selected centromedullary areas (Figure 4D). Stromal co-expression of SMA and fibronectin was located mainly in paratrabecular areas and in only a few medullary cells (Figure 3D). In contrast to tenascin, fibronectin could not be detected in single neoplastic myeloid cells (Figures 4B and 4C).
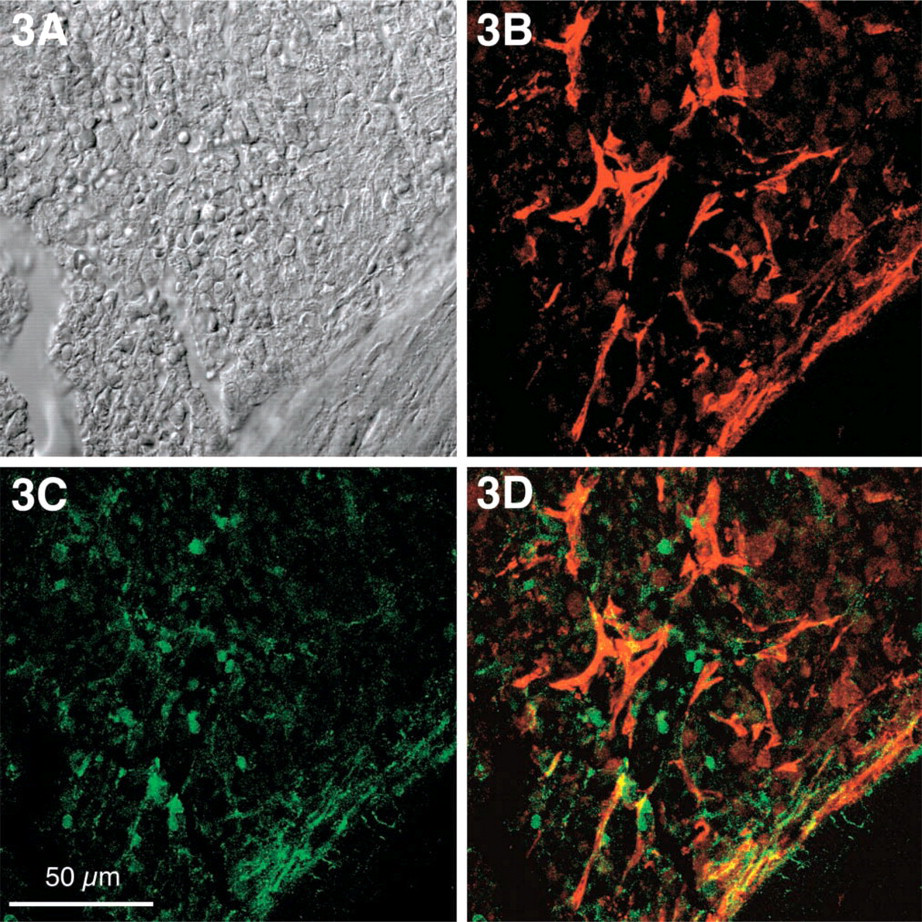
IMF in stage of moderate to severe stromal fibrosis. (
Discussion
Chronic myeloproliferative disorders (CMPDs) are clonal stem cell neoplasias characterized by the proliferation of one or more hematopoietic cell lines (Remstein et al. 2000). Medullary fibrosis is typical of idiopathic myelofibrosis but may be associated with all other subtypes of CMPD, including chronic myeloid leukemia, polycythemia vera, and essential thrombocythemia. Unlike neoplastic myeloid cells, bone marrow fibroblasts in CMPD are polyclonal, and were found to be similar to normal medullary fibro-blasts (Wang et al. 1992; Tefferi 2000). Proliferation of medullary fibroblasts and differentiation into myo-fibroblasts are believed to be induced by cytokines released by neoplastic megakaryocytes, and may also be observed at extramedullary sites colonized by clonal cells of CMPD (Castro-Malaspina et al. 1981; Remstein et al. 2000). Cytokines involved in myelofibrosis include transforming growth factor-β1 (TGF-β1), platelet-derived growth factor (PDGF), epidermal growth factor (EGF), basic fibroblast growth factor (bFGF), and calmodulin. Expression of those growth factors stimulates fibroblast activities including proliferation, migration, and deposition of various extracellular matrix proteins, i.e., collagens, fibronectin, vitronectin, laminin, and tenascin (Roberts et al. 1986; Martyre et al. 1986,1991; Kimura et al. 1988,1989; Terui et al. 1990; Pearson et al. 1988; Reilly et al. 1995; Dalley et al. 1996). Recruitment of stromal cells and induction of a myofibroblastic phenotype are mediated by granulocyte macrophage colony-stimulating factor (GMCSF) and TGF-β1, but participation of a number of other cytokines, in particular interferon-7 (7INF), tumor necrosis factor-α (TNF-α), interleukin 1 (IL-1), and interleukin 2 (IL-2) illustrate the complexity of myofibroblast differentiation (Desmoulière and Gabbiani 1994; Schürch et al. 1997).
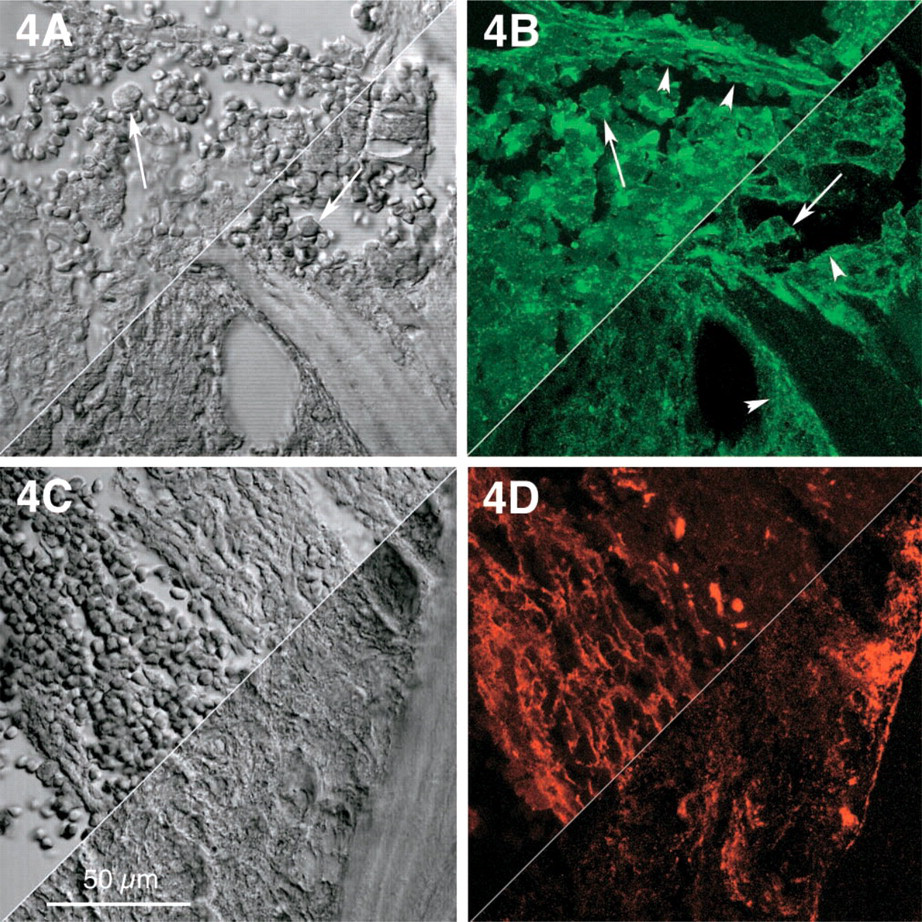
Selected centromedullary and paratrabecular areas in idiopathic myelofibrosis (
Our study, focused on the cytoskeletal SMA fibers and the two extracellular matrix proteins tenascin and fibronectin, provides further proof of a close interplay between neoplastic myeloid cells and polyclonal nonneoplastic stromal cells resembling myofibroblasts (Schmitt-Gräff et al. 1994; Hauser et al. 1995; Schürch et al. 1998; Chiquet 1999). Our results suggest that tenascin at a given stage of stromal fibrosis in the progress of CMPD may be co-expressed by both neoplastic cells of the myeloid lineage and reactive stromal cells of myofibroblastic phenotype. In stages of severe medullary fibrosis observed in IMF and occasionally in CML, however, tenascin is predominantly expressed by stromal cells and is released into the interstitial compartment, as confirmed by our results.
Tenascin has been localized in vivo and in vitro in the cytoplasm of a wide array of both mesenchymal and epithelial cells, including myofibroblasts involved in wound healing, normal and neoplastic mammary gland epithelia, oral squamous cell carcinomas, fat-storing cells of the liver, bone marrow stromal cells, and glioma cells (Van Eyken et al. 1992; Klein et al. 1993; Lightner et al. 1994; Redick and Schwarzbauer 1995; Mori et al. 1996; Seiffert et al. 1998). Proof of tenascin formation by mRNA demonstration in neoplastic myeloid cells is a subject of further study. On the other hand, phagocytosed tenascin in stromal macrophages has not been detected in our material by CD68 immunofluorescence, nor has it been reported in the literature to the best of our knowledge.
SMA and fibronectin have not been detected in myeloid cells by immunofluorescence. They reflect cytoskeletal and extracellular matrix constituents of stromal cells. Differences in the distribution pattern of tenascin and fibronectin are shared with differences in the kinetics of molecule formation. Whereas the tenascin molecule is a hexamer (termed hexabrachion) and rapidly assembled with ongoing translation before transfer to the Golgi apparatus and secretion, fibronectin molecules are gradually assembled into disulfide-bonded dimers (Redick and Schwarzbauer 1995). The rate-limiting step in the release of tenascin into the extracellular space, however, is found in the posttranslational transport from the endoplasmic reticulum to the Golgi complex, which may be less effective for slow assemblage of fibronectin dimmers (Redick and Schwarzbauer 1995). Both cytoskeletal and extracellular matrix proteins may communicate across the cell membrane, as observed in various cultured cells; this connection is receptor-mediated (Alberts et al. 1989).
The stromal bone marrow compartment includes various types of differentiated mesenchymal cells such as adipocytes, vascular smooth muscle cells, fibro-blasts, and myofibroblasts. The latter are believed to derive from a common ancestor cell and may represent various isoforms evolving on stimulation by environmental factors (Schmitt-Gräff et al. 1994; Schürch et al. 1997,1998). The cytokine microclimate reflects the functional demands in reactive processes, may mediate differentiation in cells displaying corresponding receptors, and stimulates extracellular matrix synthesis (Schürch et al. 1997,1998). Furthermore, an example of a crosstalk between the stromal and myeloid compartments may be recognized in the adhesive and mitogenic effects of tenascin on normal hematopoietic cells (Seiffert et al. 1998). Similar mechanisms appear to be effective also in neoplastic conditions (Schürch et al. 1997,1998). The documented heterogeneity in the cytoskeletal phenotype of the versatile mesenchymal stromal cell compartment is a reflection of this issue (Schmitt-Gräff et al. 1994; Schürch et al. 1997,1998).
Footnotes
Acknowledgements
Supported by the Swiss Cancer League grant no. SKL-00653–2–1998/KFS 962–9–1999.
We acknowledge Dr L. Landmann, Institute of Anatomy, University of Basle, for the complimentary access to CLSM facilities.