
Abstract
Proteinase K is widely used in methods for detection of transcripts in biological specimens by in situ hybridization (ISH). However, treatment with proteinase K hampers detection of RNA and protein simultaneously. We have developed a method for double staining of transcripts and proteins by ISH and IHC staining in imaginal discs and embryos of Drosophila. Instead of treatment with proteinase K, samples are treated with ethanol plus xylene and with acetone. Acetone renders cell membranes permeable to probes and antibodies without damaging tissue integrity, whereas treatment with proteinase K sometimes damages tissues. Treatment of samples with acetone allows hybridization of probe with transcripts in tissue. It is also effective for immunological staining of samples after ISH with a riboprobe. Thus, our method allows detection not only of transcripts but also of specific proteins in relatively intact single samples. (
W
Here we describe an improved method for ISH and its application to detection of RNA and protein simultaneously in imaginal discs and embryos of Drosophila. We omit the usual initial treatment with proteinase K and treat samples with acetone instead. Treatment of specimens with acetone allows successful immunological staining of tissues. The use of acetone maintains the integrity of tissues without the major damage that is sometimes caused by treatment with proteinase K. Acetone also renders cell membranes permeable to probes and antibodies without any damage to tissue integrity, allowing both ISH and immunological staining.
Dissection and Fixation
Larvae at the late third-instar stage were dissected and the central nervous system (CNS) and imaginal disc complex were transferred to ice-cold paraformaldehyde (PFA; 4%). Samples were fixed, washed with methanol, and stored at −20C. Embryos were dechorionated in 50% commercial chlorine bleach, rinsed with distilled water, washed with a solution of 0.07% NaCl plus 0.01% Triton X-100, and then fixed in 4% PFA under heptane at room temperature (RT) for 20 min. Embryos were then devitellinized with cold methanol and stored at −20C. For immunological staining and detection of fluorescence, 4% PFA was replaced by 4% formaldehyde (FA).
Initial Treatment of Samples
Fixed samples were treated by Method A or Method B before hybridization, as described below. All steps were performed on ice unless otherwise specified. In cases of detection with horseradish peroxidase, samples were incubated in methanol that contained 0.3% (v/v) hydrogen peroxide at RT for 60 min before the initial treatment.
Method A without Proteinase K
Fixed samples were washed in methanol and then rehydrated by successive treatments with a graded methanol series (80%, 50%, 25% v/v in H2O) and PTwx (PBS plus 0.1% Tween-20 and 0.1% Triton X-100) for 5 min each. Then samples were fixed again in PBS that contained PFA (4%) for 20 min and, finally, they were washed three times in PTwx for 5 min each.
Method A with Proteinase K
This protocol was similar to the method described above except for the incubation with proteinase K. Samples were treated with proteinase K (20 μg/ml) for 5 min at 24C and then incubated for 5 min in PBS that contained glycine (10 mg/ml). They were then washed and fixed as described above.
Method B
Fixed samples were washed twice with ethanol. Then they were incubated in a mixture of xylene and ethanol (1:1 v/v) for 60 min, washed twice in ethanol for 5 min each, and rehydrated by immersion in a graded methanol (80%, 50%, 25% v/v in H2O) and H2O for 5 min each. After treatment with acetone (80%) at −20C for 10 min, samples were washed twice with PTwx for 5 min each and fixed again in PBS that contained PFA (4%) for 20 min. Finally, they were washed in PTwx as described above.
Hybridization and Detection
After treatment of samples by the various methods described above, hybridization and washing were performed as described elsewhere (Nagaso et al. 1999; Nikaido et al. 1999). RNA probes for transcripts were synthesized in vitro with T7 RNA polymerase (Roche Diagnostics; Tokyo, Japan) using plasmid DNA that encoded the dpp (Padgett et al. 1987), wg (Siegfried et al. 1994), hid (Grether et al. 1995), Ser (Thomas et al. 1991) and stg (Edgar and O'Farrell 1989) genes. After incubation in a blocking buffer that contained 100 mM maleic acid (pH 7.5), 150 mM NaCl, and 2% blocking reagent (Roche Diagnostics), the samples were incubated overnight in blocking buffer supplemented with 1000-fold diluted DIG-specific antibodies conjugated with alkaline phosphatase. Color was developed by incubation in alkaline phosphatase buffer [100 mM Tris-HCl (pH 9.5), 100 mM NaCl, 0.3% Tween-20] supplemented with 5 mM levamisole and 4-nitroblue tetrazolium chloride-5-bromo-4-chloro-3-indolyl-phosphate (NBT-BCIP) as substrate. For detection of fluorescence, color was developed by incubation in 1 × TET buffer [10 mM Tris-HCl (pH 8.0), 2 mM EDTA, 0.3% Tween-20] supplemented with FastRed (Roche Diagnostics Co.).
In Situ Hybridization and Immunological Staining
Samples were allowed to react simultaneously with primary antibodies and antibodies against DIG (Roche Diagnostics), as described above. The following primary antibodies were used: antibodies specific for Cut (2B10; Blochlinger et al. 1990), Elav (9F8A9; O'Neill et al. 1994), En (4D9; Patel et al. 1989), and wg (4D4; Siegfried et al. 1994), and all were supplied by the Developmental Studies Hybridoma Bank (Iowa City, IA). The following secondary antibodies were used: antibodies specific for mouse IgG, conjugated with Cy5 (Jackson Immuno Research; West Grove, PA), with HRP (Santa Cruz Biotechnology; Santa Cruz, CA) or with biotin (Jackson Immuno Research). After washing, color was developed by reaction with a solution of 3,3′-diaminobenzidine (DAB; Nitirei, Tokyo, Japan) and amplified with an ABC kit (Vector Laboratories; Burlingame, CA). After the immunological staining, reactions for detection of ISH of transcripts with alkaline phosphatase were performed as described above.
For ISH in Drosophila, we modified methods used for zebrafish that had been described previously (Nagaso et al. 1999; Nikaido et al. 1999). We first performed a comparative study of methods for detection in situ of transcripts of the wingless (wg) and decapentaplegic (dpp) genes in the wing imaginal discs of Drosophila (Padgett et al. 1987; Siegfried et al. 1994). The wg gene is expressed at the boundary of the dorsal and ventral compartments of the presumptive wing pouch (Baker 1988a, b; Neumann and Cohen 1996). By contrast, the dpp gene is expressed at the boundary of the apical and proximal compartments of wing imaginal discs (de Celis 1997).
We used two protocols, i.e., Method A and Method B, for ISH. In Method A, we compared signals specific for dpp and wg in samples that had been fixed in formaldehyde and methanol with and without initial treatment of samples with proteinase K. Nagaso et al. (1999) used Method A, without proteinase K, to detect transcripts in early embryos of zebrafish by ISH and found that it did not generate any background signals. Moreover, Knirr et al. (1999) reported that Method A with proteinase K was effective for in situ detection of transcripts in imaginal discs of Drosophila embryos. However, in imaginal discs, extended incubation for visualization of sites of alkaline phosphatase activity resulted in ectopic staining as background noise. In our analysis, signals associated with expression of the dpp (Figure 1A) and wg (Figure 1B) genes were widely distributed over the entire wing pouch but were sometimes rather weak (Figure 1A, arrow) or absent in the presumptive notum (data not shown). In an attempt to reduce the background staining in the wing pouch and to enhance signals in the notum, we tested the effect of inclusion of digestion with proteinase K in Method A. In this case, background signals in the entire wing pouch were reduced and expression of the dpp (Figure 1C) and wg (Figure 1D) genes was clearly detectable in pouches of wing discs, as described elsewhere (Baker 1988a, b; Neumann and Cohen 1996; de Celis 1997). However, treatment with proteinase K resulted, on occasion, in weakened signals in the notum and in damage to discs (data not shown). To solve these problems, we developed a novel method for ISH using successive treatments with organic solvents, i.e., ethanol, xylene and acetone, which we refer to as Method B.
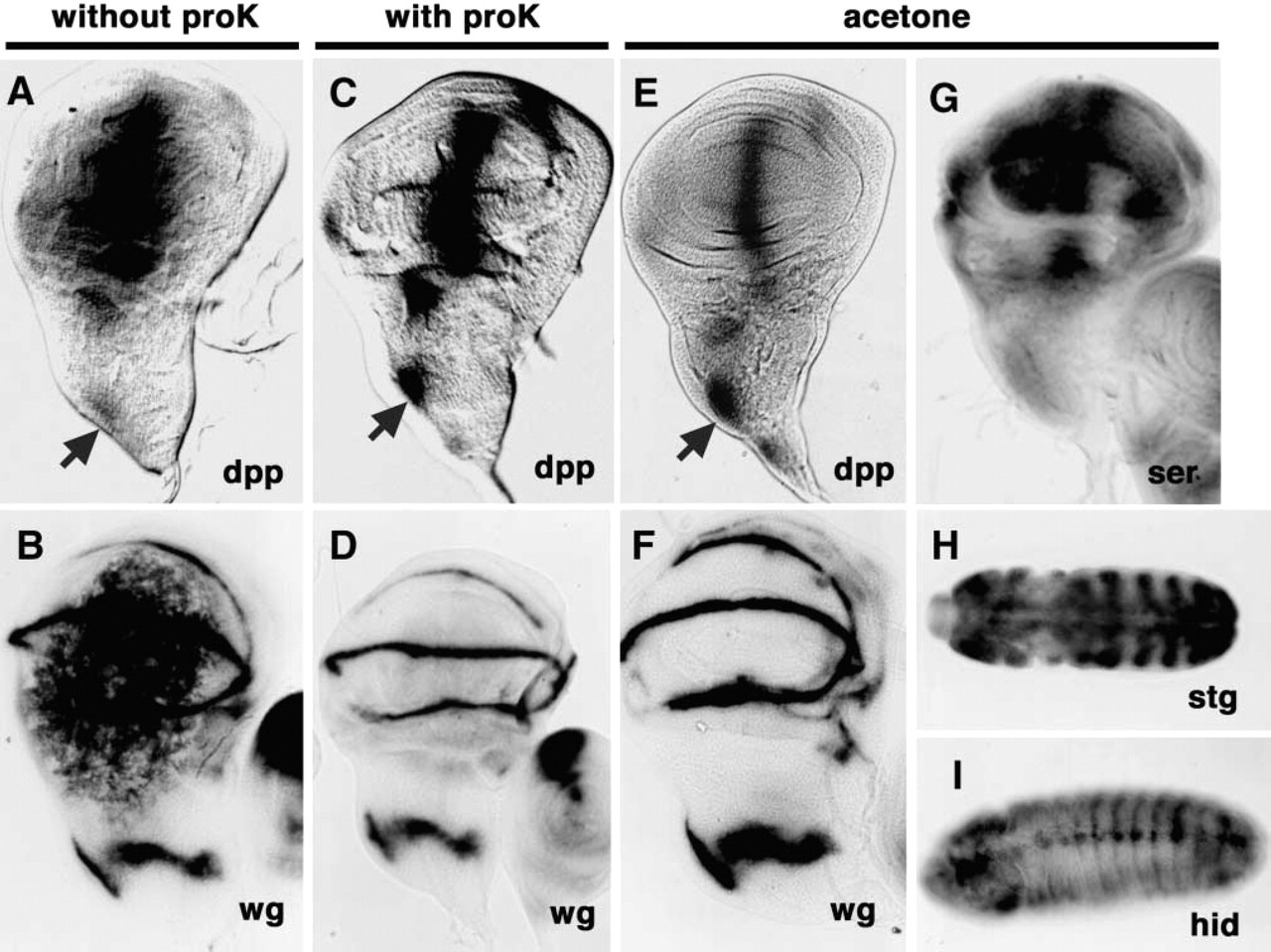
Effects of treatment with acetone on ISH. (
In Method B, samples fixed in formaldehyde and methanol were treated with acetone for 10 min at −20C instead of being treated with proteinase K. The background signals were significantly reduced but did not disappear completely. To reduce the background signals still further, we treated samples with ethanol and xylene, which has been reported to reduce the background signals when used in combined with proteinase K (Harlow and Lane 1999). Thus, the treatment of samples with ethanol plus xylene for 60 min and then with acetone for 10 min at −20C was effective for ISH of transcripts without damaging tissue integrity. The signals specific for wg transcripts in the notum region and the outer-ring region that surrounded the wing pouch were as clear when we used Method B as when we used Method A with proteinase K (Figures 1D and 1F). We obtained similar results in the case of dpp transcripts (Figures 1C and 1E). Therefore, the use of Method B appears optimal for in situ detection of transcripts in the imaginal discs of Drosophila larvae because treatment with ethanol plus xylene and then with acetone yielded signals specific for transcripts of dpp and wg. We also examined signals specific for transcripts of other genes such as string (stg) (Edgar and O'Farrell 1989; Edgar et al. 1994), hid (Grether et al. 1995; Kurada and White 1998) and Serrate (Ser) (Thomas et al. 1991) in the imaginal discs by Method B as well as in Drosophila embryo, and in each case we detected specific signals (Figures 1G–1I).
Immunological methods designed to allow detection of RNA and protein simultaneously have recently been developed (Saunders and Cohen 1999). Because treatment of samples with proteinase K can result in loss of antigenicity of proteins as a result of nonspecific degradation, problems can arise in attempts at double staining of transcripts and proteins. Therefore, we compared Method A with Method B in terms of the efficiency of subsequent immunological staining. We examined specific signals caused by binding of antibodies against Cut (Blochlinger et al. 1990, 1993; Jacobsen et al. 1998), Elav (O'Neill et al. 1994), En (Patel et al. 1989, 1994), and Wg (Brook and Cohen 1996; Siegfried et al. 1994) in imaginal discs and embryos after samples had been prepared either by Method A or by Method B. All signals were detected after preparation by Method B (Figures 2B, 2C, 2G–2I), as well as by Method A without proteinase K (data not shown). As expected, Method A with proteinase K sometimes resulted in the degradation of Cut, Elav, and En proteins in embryos (Figures 2D–2F). In imaginal discs, Elav was fully degraded (data not shown), while Cut (Figure 2A) and En (data not shown) were degraded to a lesser extent. In the case of Wg protein, there was no difference in terms of signals between Method A and Method B (Figure 3D; and data not shown). Therefore, signals due to the respective proteins could be detected at high efficiency after samples had been permeabilized by Method B. Treatment with ethanol plus xylene and acetone was able to replace treatment with proteinase K in the preparation of samples for ISH and, moreover, it abolished problems associated with nonspecific degradation of samples caused by Method A, with or without proteinase K. Treatment with ethanol, with xylene, or with acetone alone could not replace treatment with the combination of organic solvents. We also examined various conditions of acetone treatment, such as concentration of acetone, temperature for incubation, and incubation period. The conditions of 50% acetone at either −20C or 4C and for 30 min or 60 min are not optimized. We found that the condition of 80% acetone at −20C for 10 min is optimal.
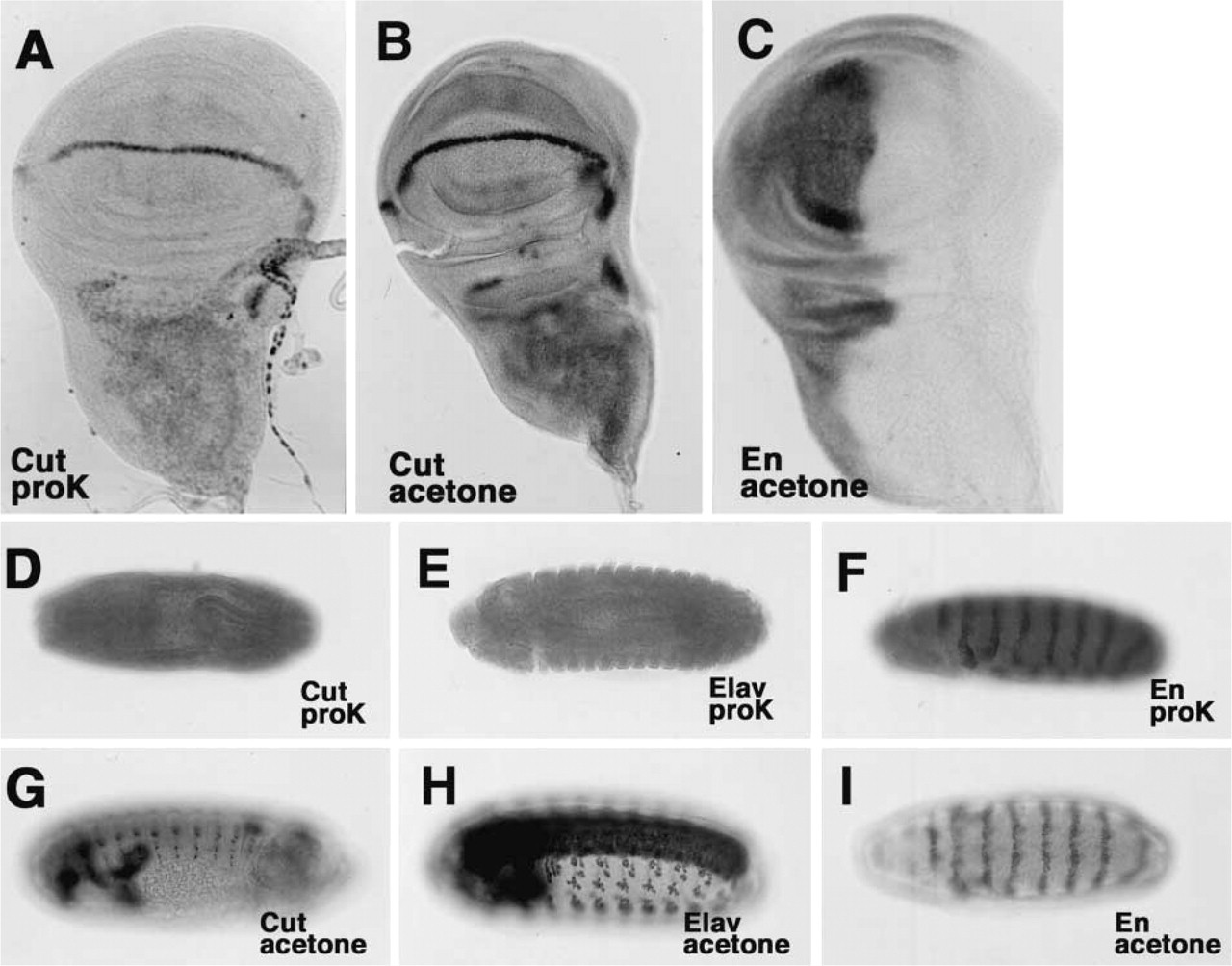
Treatment with acetone is critical for immunological staining. (
Finally, we examined the immunological staining of Cut, En, and Wg proteins after ISH of transcripts (Figure 3). We successfully detected, for example, signals specific for the En protein and the wg transcript in both discs and embryos (Figures 3A, 3B and 3B′). Weaker signals, such as those due to the Cut protein in embryos, could be amplified with an ABC kit (Figures 3C and 3C′). In addition, recently developed methods for detection using fluorescent substrates and amplified systems, such as Fast Red (Jowett and Yan 1996), could be applied without problems after preparation of samples by Method B (Figures 3D–3F). Thus, Method B was effective for in situ immunological detection of both RNA and protein because samples remained relatively intact. In conclusion, our improved method for ISH using acetone in place of proteinase K is simple and rapid and appears optimal for detection of transcripts and proteins in a single specimen without significant damage to tissue integrity.
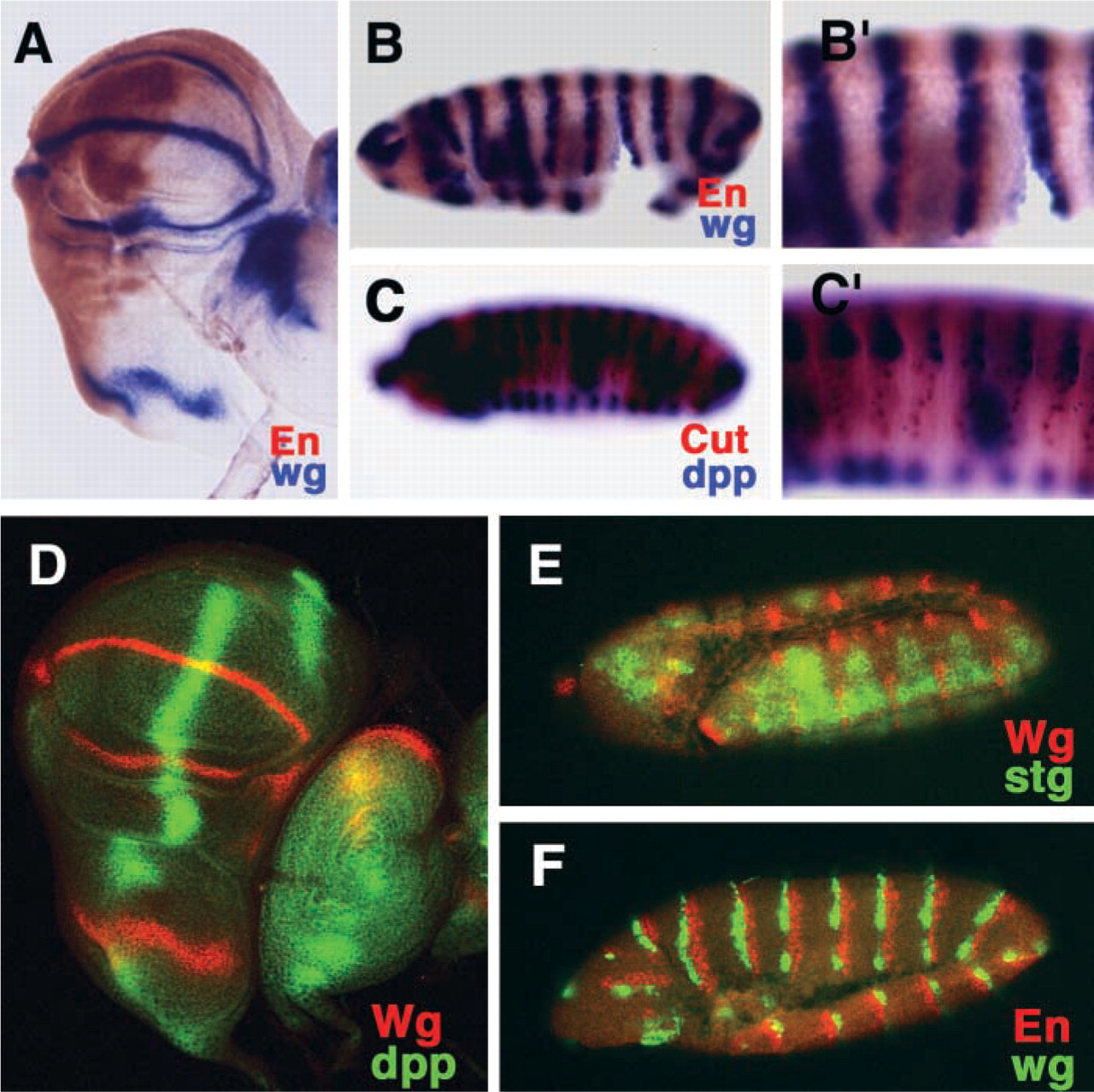
Double staining for the detection of specific proteins and transcripts. (
Footnotes
Acknowledgments
We thank A. M. Körner for critical reading of the manuscript.
Supported by grants from RIKEN for Bioresource Research Projects, from the Uehara Memorial Foundation, and from the Special Coordination Funds of the Ministry of Education, Culture, Sports, Science and Technology of Japan (to KKY).