
Abstract
We have developed a branched DNA in situ hybridization (bDNA ISH) method for detection of human papillomavirus (HPV) DNA in whole cells. Using human cervical cancer cell lines with known copies of HPV DNA, we show that the bDNA ISH method is highly sensitive, detecting as few as one or two copies of HPV DNA per cell. By modifying sample pretreatment, viral mRNA or DNA sequences can be detected using the same set of oligonucleotide probes. In experiments performed on mixed populations of cells, the bDNA ISH method is highly specific and can distinguish cells with HPV-16 from cells with HPV-18 DNA. Furthermore, we demonstrate that the bDNA ISH method provides precise localization, yielding positive signals retained within the subcellular compartments in which the target nucleic acid sequences are localized. As an effective and convenient means for nucleic acid detection, the bDNA ISH method is applicable to the detection of cancers and infectious agents.
(
Keywords
I
Branched DNA (bDNA) signal amplification technology has been used extensively in a microwell format to detect and quantify specific nucleic acid sequences. Inherently quantitative and highly reproducible, bDNA technology utilizes nonradioactive synthetic oligonucleotide probes and can readily be applied to the detection of any nucleic acid target for which a sequence is known. A number of bDNA assays have been developed for quantification of viral nucleic acids, including human immunodeficiency virus type 1 (HIV-1) RNA (Kern et al. 1996), simian immunodeficiency virus (SIV) RNA (Sodora et al. 1998), hepatitis B virus (HBV) DNA (Hendricks et al. 1995), hepatitis C virus (HCV) RNA (Detmer et al. 1996), hepatitis G virus (HGV) RNA (Brandhagen et al. 1999), and cytomegalovirus (CMV) DNA (Chernoff et al. 1997; Pellegrin et al. 2000). More recently, bDNA technology has been used to detect and measure the expression of cellular mRNAs, including cytokines (Breen et al. 1997; Shen et al. 1998), progesterone and estrogen receptors (Nargessi et al. 1998a,b), insulin (Wang et al. 1997), glucokinase (Cabrera–Valladares et al. 1999), c-fos (Shyamala et al. 1999), aP2 (Burris et al. 1999), cytochrome P450 (Hartley and Klaassen 2000), and uncoupling proteins (Zhou et al. 2000). Although these bDNA assays were developed to measure nucleic acids in serum, plasma, or cell lysates, other studies have shown that it is possible to adapt bDNA technology to an ISH format for detection of mRNA (Cao et al. 1998; Antao et al. 2000).
Here we report the experimental results obtained after development of a bDNA ISH method for the detection of human papillomavirus (HPV) DNA and mRNA in whole cells. The bDNA ISH method utilizes a series of non-isotopic oligodeoxyribonucleotide probes, hybridized sequentially, to generate chromogenic and fluorescent signals. By modifying conditions of cell pretreatment, either mRNA or DNA can be detected with the same set of probes. Using human cervical cancer cell lines that are well characterized with regard to HPV genotype and number of integrated HPV genomes, we evaluate the sensitivity and specificity of the bDNA ISH method. Furthermore, we assess whether signals generated by the bDNA ISH method are retained in the subcellular compartments in which the target nucleic acid sequences are localized.
Materials and Methods
Cell Culture
Human cervical cancer cell lines were obtained from the American Type Cell Culture Collection (ATCC; Manassas, VA). Cell lines positive for HPV-16 or HPV-18 included HeLa, CaSki, and SiHa, which are well characterized with regard to genotype and number of integrated HPV genomes (Yee et al. 1985; Baker et al. 1987; Mincheva et al. 1987; Siadat-Pajouh et al. 1994; Plummer et al. 1998; Meissner 1999). Controls included the HPV-39-positive ME180 and the HPV-negative C33A and HT3 cell lines. Cells were grown either in flasks or on chamber slides coated with 10 μg/ml poly-
Oligonucleotide Probes
A total of 26 HPV-16-specific and 32 HPV-18-specific DNA degenerate oligonucleotide target probes were designed to cover 90% of the E6 and E7 regions of the HPV genome. These probes were generated using the ProbeDesigner Software (Bayer Diagnostics; Emeryville, CA) (Bushnell et al. 1999) with a constant melting temperature of 63 ± 2C. Final probes were selected after screening for possible interactions with 39 other HPV genotypes as well as human genomic DNA sequences by using HybSimulator software (Advanced Gene Computing Technologies; Irvine, CA) and Blast2 software (National Center for Biotechnology Information; National Library of Medicine, NIH, Bethesda, MD). Other DNA oligonucleotide probes, which are part of the signal amplification system and include preamplifier, amplifier, and alkaline phosphatase (AP)-conjugated label probes, have been described in detail (Collins et al. 1997). To reduce potential nonspecific hybridization, non-natural nucleotides 5-methyl-2′-deoxyisocytidine (isoC) and 2′-deoxyisoguanosine (isoG) were included in the target, preamplifier, amplifier, and AP-conjugated label probes. Although non-natural nucleotides have recently been incorporated into bDNA technology, specific and sensitive detection of target nucleic acid sequences is routinely achieved using probes without non-natural nucleotides (Urdea and Wuestehube 2000).
bDNA ISH for DNA Detection
Harvested cells were fixed with 4% formaldehyde in PBS (0.01 M phosphate buffer, pH 7.5) for 30 min on ice, pipetted into double-spotted cytospin funnels (Shandon; Pittsburgh, PA), and centrifuged for 6 min at 1500 rpm in a cytospin centrifuge. The cells were dehydrated on slides through a graded ethanol series, air-dried at RT for 10 min, and stored at −80C for up to 2 weeks. For pretreatment, cells were rehydrated on slides through a graded ethanol series and washed in PBS. Slides were incubated at 37C for 1 hr in 40 μg/ml RNase in 2 × SSC (1 × SSC is 0.15 M NaCl, 0.015 M Na-citrate), and washed in PBS, before incubation in 7.5–10 μg/ml proteinase K in PBS at 37C for 10 min. Cells were then dehydrated and air-dried at RT for 10 min.
After pretreatment, cells were incubated with a prehybridization solution, hybridized with a set of oligonucleotide target probes (described above), and then hybridized with a series of oligonucleotide probes for signal amplification (Figure 1). As a prehybridization step, each spot on the slide was incubated with 150 μl hybridization Solution A [3 × SSC, 50% formamide, 10% dextran sulfate (MW 500,000), 0.2% casein, 10 μg/ml poly A, and 100 μg/ml denatured salmon sperm DNA] at RT for 30 min. Cells were denatured in a humidity chamber (Hybaid Omnislide instrument; Phenix Research Products, Hayward, CA) at 92C for 10 min and then cooled at RT for 5 min. Cells were incubated in 100 μl of hybridization Solution A containing 0.6 pmole HPV-specific target probes at 40C for 3 hr in a humidified chamber. Slides were washed at RT with a decreasing series of SSC buffers containing 0.0025% Brij-35 detergent (Surfact-Amps 35; Pierce, Rockford, IL) for 1–2 min each in 2 × SSC, 0.2 × SSC, 0.1 × SSC, and 2 × SSC. Cells were then incubated with 100 μl hybridization Solution B (5 × SSC, 0.1–0.3% SDS, 10% dextran sulfate, 1 mM ZnCl2, and 10 mM MgCl2) containing 90 fmoles preamplifier at 55C for 25 min in the humidity chamber. Slides were washed twice in 0.1 × SSC, 1 mM EDTA for 1 min and 4 min, respectively, and then incubated in 100 μl hybridization Solution B containing 90 fmoles amplifier at 55C for 25 min in the humidity chamber. Slides were washed twice in 0.1 × SSC, 1 mM EDTA for 1 min and 4 min, respectively, and then incubated in 100 μl hybridization Solution B containing 90 fmoles AP-conjugated label probe at 55C for 15 min in the humidity chamber. After washing in 100 mM Tris, pH 8.0, containing 0.1% Brij-35, 1 mM ZnCl2, and 10 mM MgCl2 at RT for 5 min, cells were incubated in 50 μl buffered AP substrate (Fast Red, #K597; DAKO, Carpinteria, CA) at RT for 10 min. Slides were counterstained with either Gills hematoxylin or 0.0001% bisbenzimide. Slides were mounted with Ultramount (DAKO), Permount, or 75% glycerol and stored at RT. Slides were viewed using a Nikon E800 fluorescence microscope with an FITC or triple bandpass filter or a ×60 brightfield objective, and images were captured using an Optronics cooled 3-chip color CCD camera. Fluorescent or chromogenic images were captured using Image Probe Software (Media Cybernetics; Silver Springs, MD). Figures were generated using Adobe Photoshop 3.0 and printed using a Sony Digital Printer.
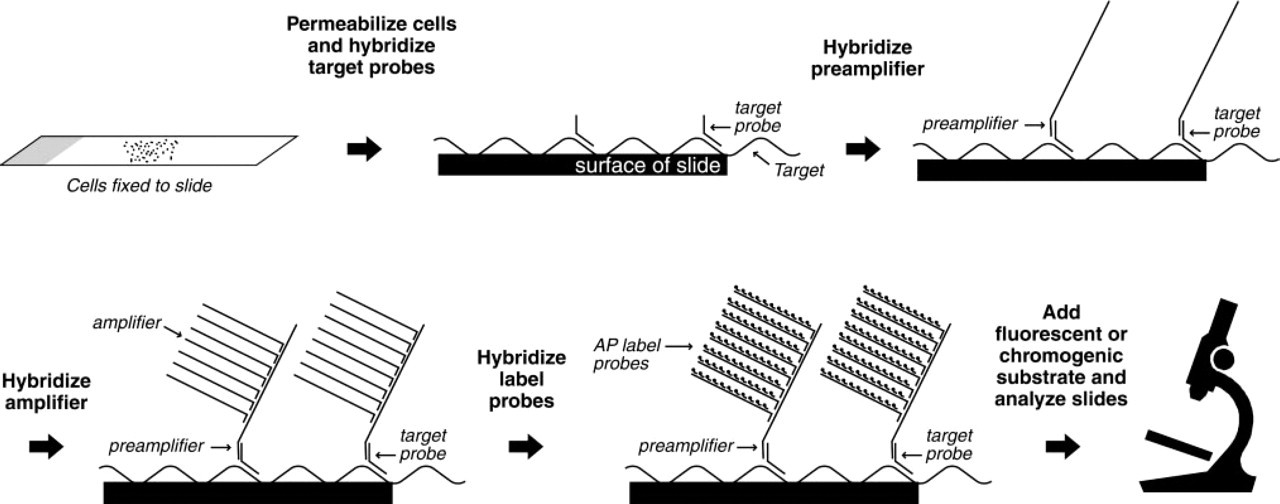
Schematic depiction of the bDNA ISH method.
For the cell mixing experiments, HeLa cells were labeled with the fluorescent cell tracer carboxyfluorescein diacetate, succinimidyl ester (CFDA SE; Molecular Probes, Eugene, OR) at 37C for 15 min in PBS containing 5 μM CFDA SE. After washing away excess CFDA SE, the cells were incubated at 37C for an additional 30 min in PBS. Cells were harvested, fixed in 4% formaldehyde in PBS, mixed with unlabeled formaldehyde-fixed CaSki cells, and then cell mixtures were spun onto cytospin slides and assessed for DNA targets, as noted above.
bDNA ISH for RNA Detection
Cells grown on chamber slides were fixed with 4% formaldehyde in PBS for 30 min at RT, treated with 10 μg/ml proteinase K in PBS for 10 min at RT, and then washed twice for 5 min in PBS. Samples were incubated at 40C for 3 hr with 1 pmole HPV-specific target probes in target probe buffer (6 × SSC, 25% formamide, 0.2% Brij-35, and 0.2% casein) and then washed with the same decreasing series of SSC buffers as was used in the bDNA ISH protocol for DNA detection. Similarly, the preamplifier, amplifier, and AP-conjugated probe hybridization and wash conditions were the same as that for the bDNA ISH protocol for DNA detection. AP substrate was added and slides were incubated at RT for 4 min. After stopping the reaction by washing in PBS, samples were postfixed in 4% formaldehyde in PBS for 5 min at RT and then counterstained for 40 sec with either hematoxylin or bisbenzimide. Slides were viewed and images were generated and printed as described above.
Results
Sensitivity and Specificity of bDNA ISH for HPV DNA Detection
The bDNA ISH method is illustrated in Figure 1. After fixation and permeabilization of cells, the target DNA or RNA is hybridized to a series of synthetic oligonucleotide probes. First, a set of target probes is hybridized to the target nucleic acid molecules in the cell. Preamplifier molecules are hybridized to the target probes, providing a bridge for the hybridization of amplifier molecules. Amplification of the signal is accomplished by the binding of up to 14 amplifier molecules to each preamplifier and of up to 196 AP-conjugated label probes to each target probe. Addition of Fast Red AP substrate results in the deposition of a red reaction product in the vicinity of the target nucleic acid that can be visualized with standard optical light or fluorescent microscopy.
The results shown in Figure 2 illustrate the effectiveness of the bDNA ISH method for detection of viral DNA and gene expression. The HPV-16 genome was detected in CaSki cells (Figure 2A) and SiHa cells (Figure 2B), which contain 400–600 and 1–2 copies of HPV-16 DNA, respectively. No signal was detected with HPV-16 probes in cells lacking HPV-16 DNA, including HeLa (Figure 2C) and C33a cells (Figure 2D). On hybridization with HPV-18 target probes, positive signal detection was observed in HeLa cells (Figure 2G), which contain 10–50 copies of HPV-18 DNA. No signal was detected with HPV-18 probes in cells lacking HPV-18 DNA, including CaSki (Figure 2E), SiHa (Figure 2F), and C33a cells (Figure 2H). No signal was detected with either HPV-16 or HPV-18 target probes in the HPV-negative HT3 cell line or the ME180 cell line, which harbors a DNA sequence similar to that of HPV-39 (not shown). These results show that one to two copies of HPV-16 DNA are detected in SiHa cells using the bDNA ISH method. Furthermore, the target probes used for bDNA ISH can distinguish between HPV-16 and HPV-18 genomic sequences. Although in some experiments 100% of cells were positive for HPV-16 DNA (CaSki and SiHa) or HPV-18 DNA (HeLa) detection, 70–90% of HPV-positive cells per slide is a conservative estimate of what was observed on a routine basis.
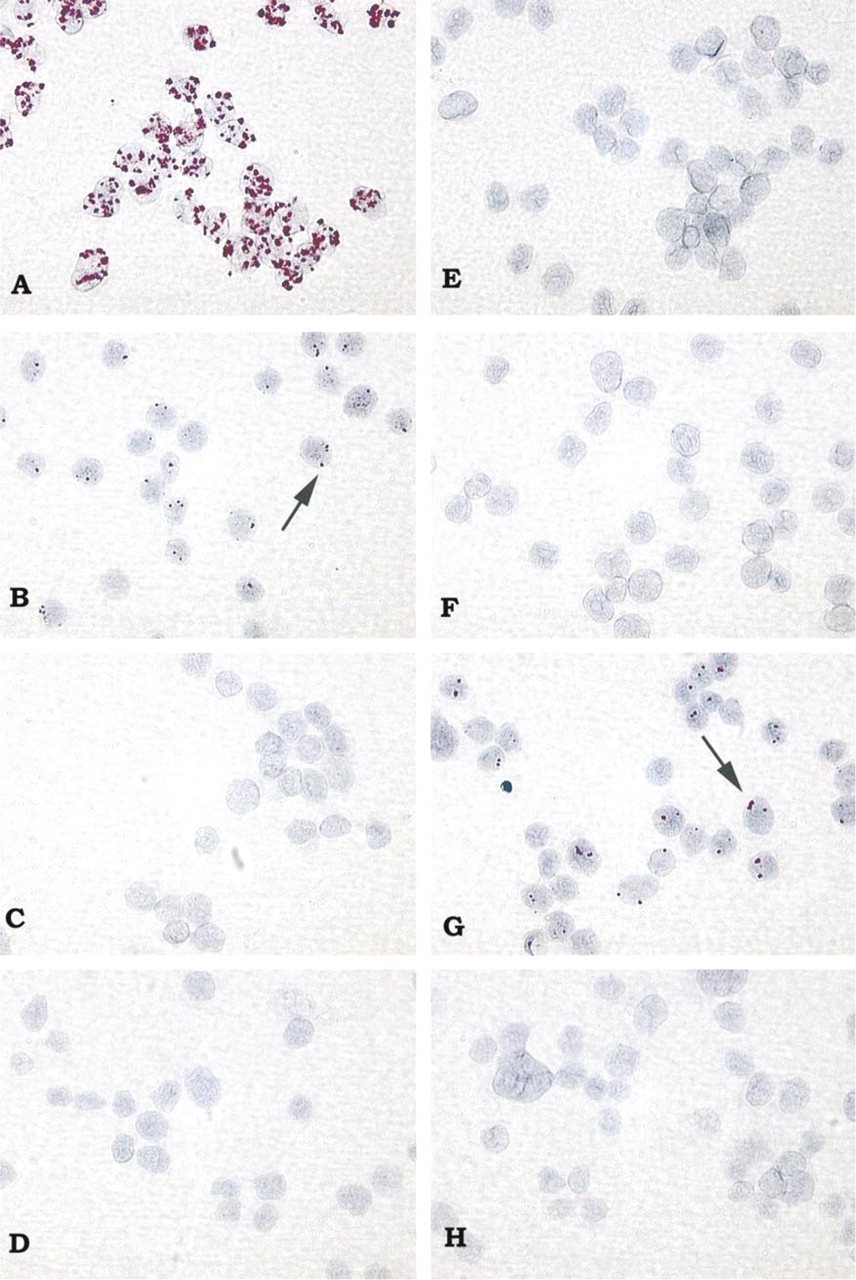
Sensitivity of bDNA ISH for detection of HPV DNA in human cervical carcinoma cell lines. Hybridization of HPV-16 probes results in deposition of Fast Red–AP reaction product in CaSki cells containing 400–600 copies HPV-16 DNA per cell (
A number of additional controls were performed to determine whether the signals observed were specific for HPV DNA targets (not shown). No signal was observed when nonspecific target probes were used or when HPV-16 or HPV-18 target probes, amplifier, or AP-conjugated label probes were omitted from the bDNA ISH method. Omission of the proteinase K digestion or DNA denaturation steps or treatment of cells with DNase also resulted in a loss of signal. As a further control for the DNase digestion experiments, the RNase treatment step was omitted to confirm that this loss in signal was not due to DNase degradation of the DNA oligonucleotide probes. Under these conditions, signal was again detected, indicating that the DNA oligonucleotide probes were not degraded and thus were capable of binding to HPV RNA targets.
Genotype-specific Detection of HPV DNA by bDNA ISH
Mixed cell populations were used to further assess the specificity of the bDNA ISH method for HPV DNA detection. Mixed cell samples were composed of unlabeled CaSki cells (containing 400–600 copies of HPV-16 DNA) and CFDA-labeled HeLa cells (containing 10–50 copies of HPV-18 DNA). As shown in Figure 3, hybridization with HPV-16 target probes yielded signal detection only in HPV-16-infected CaSki cells and not in HeLa cells (Figure 3A and 3C). HeLa cells surrounded by positively stained CaSki cells sometimes appeared to be partly stained, but this likely is an artifact from overlapping CaSki and HeLa cells (indicated by arrow in Figure 3A). Likewise, hybridization with HPV-18 target probes yielded signal detection only in HPV-18-infected HeLa cells and not in CaSki cells (Figures 3B and 3D). Although the vast majority of HeLa cells were well labeled with the CFDA SE cell tracer, a few cells were faintly labeled (Figure 3D, cell at middle bottom). As expected, greater signal intensity (i.e., larger number and size of spots) was observed for HPV-16 DNA detection in CaSki cells compared to HPV-18 DNA detection in HeLa cells. This difference in signal intensity is likely a reflection of the higher HPV DNA copy number present in CaSki cells (400–600 HPV-16 DNA copies/cell) compared to HeLa cells (10–50 HPV-18 DNA copies/cell). These results demonstrate that, in a mixed population of cells, the bDNA ISH method can distinguish cells with HPV-16 DNA from cells with HPV-18 DNA. Because signals are retained within the appropriate cell types, there is no diffusion of the AP–Fast Red reaction product (Speel et al. 1992) from one positive cell type to another negative cell type within the same sample. Moreover, even low-abundance viral targets can be readily detected with the bDNA ISH method without interference from nonspecific hybridization to related viral sequences or other sequences.
Subcellular Co-localization of Signal and Target Nucleic Acids with bDNA ISH
Because subcellular compartments are more easily visualized in cells cultured on chamber slides, HeLa cells grown on chamber slides were used to assess whether the bDNA ISH method could detect the subcellular distribution of HPV mRNA and HPV DNA. As shown in Figure 4, hybridization of HeLa cells (prepared for RNA detection) with HPV-18 target probes resulted in detection of HPV-18 mRNA mainly in the cytoplasm (Figure 4A). Although mRNA detected in the cytosol appears to overlap with cell nuclei when whole cells are viewed through a microscope, this is consistent with the cell nucleus being naturally surrounded by cytosol within cells. No signal was observed in these same cells incubated with HPV-16 target probes (Figure 4B). In contrast, hybridization of HeLa cells (prepared for DNA detection) with HPV-18 target probes resulted in the detection of HPV-18 DNA in HeLa cell nuclei (Figure 4C). No signal was observed in these same cells incubated with HPV-16 target probes (Figure 4D). These results demonstrate that the signal observed with bDNA ISH is retained in the subcellular compartment known to contain the nucleic acid target: viral mRNA is detected in cytosol, whereas viral DNA is detected in cell nuclei. In other words, the target and signal are co-localized with the bDNA ISH method.
Discussion
The bDNA ISH method described here provides a rapid, sensitive, and reproducible means for detection of specific DNA and mRNA sequences in various cell types. Our results show that the sensitivity of the bDNA ISH method is sufficient to detect relatively low-abundance targets, as few as one or two copies of HPV-16 DNA in SiHa cells. Our results also demonstrate that the bDNA ISH method is highly specific and provides subcellular localization of the target sequence. In mixed cell population experiments, signals were detected only in the appropriate cell types, indicating that there was no diffusion of AP–Fast Red reaction product from one cell type to another. Moreover, signals obtained with bDNA ISH are retained within the cellular compartment in which the target sequence is localized. Subcellular localization experiments with HPV-18-infected HeLa cells showed that viral mRNA was predominantly detected in the cytoplasm, whereas viral DNA was detected in cell nuclei.
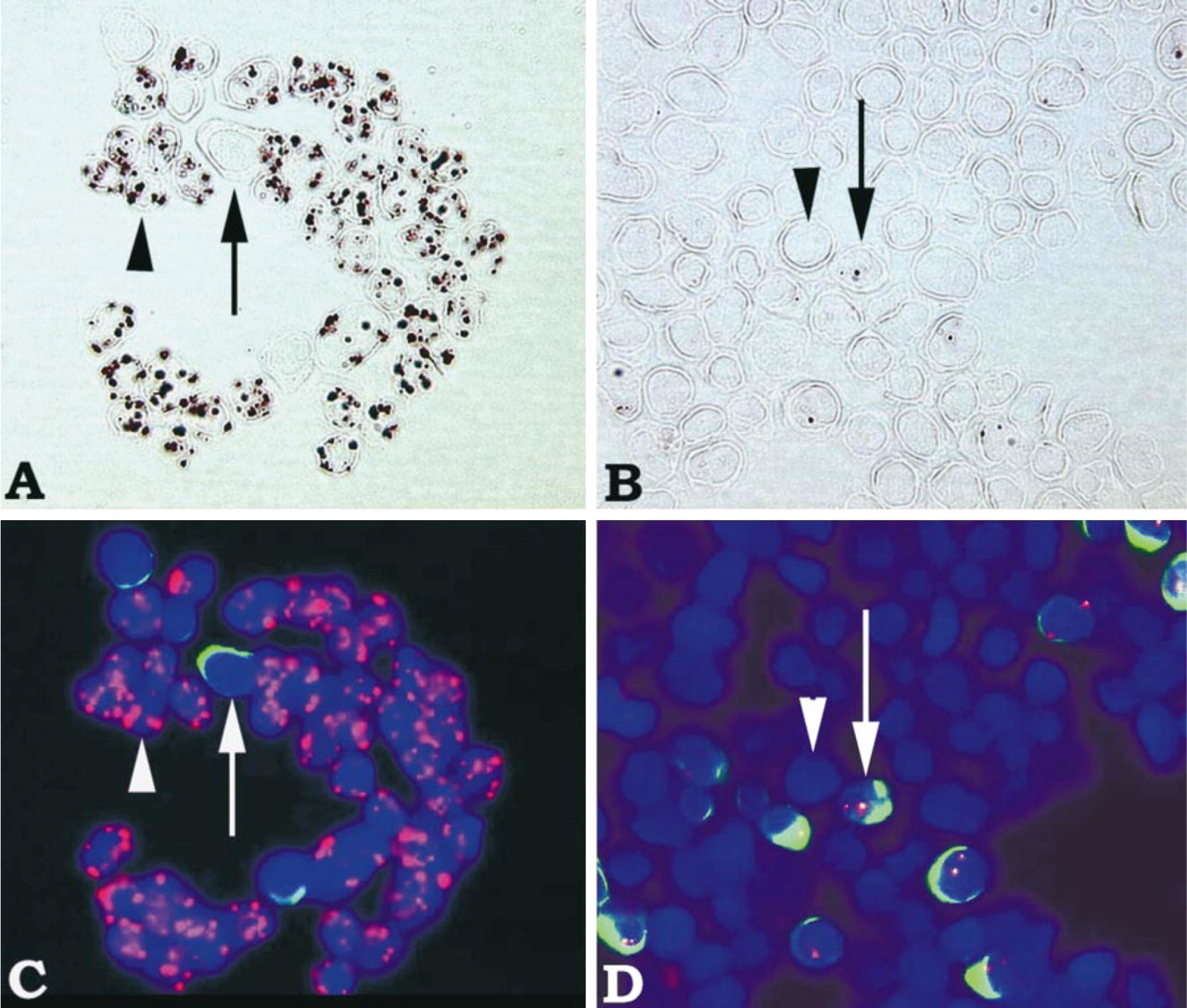
Genotype-specific detection of HPV DNA by bDNA ISH in mixed samples of HPV-18-positive HeLa cells and HPV-16-positive CaSki cells. For ready identification of cell types, HeLa cells were labeled with CFDA SE cell tracer. In all images, HeLa cells are indicated by arrows and CaSki cells are indicated by arrowheads. In fluorescent images (
The bDNA ISH method described here works well, with very little modification, when different target probe sets and different cell and tissue types are used. We have reproducibly detected single-copy HIV-1 DNA targets in HIV-positive 8E5 cells and two copies of the heterogeneous nuclear ribonuclear protein gene in CaSki and 8E5 cell lines (unpublished work). Although in the present study we have limited our focus to HPV detection in whole cells, in other experiments we have detected HPV in paraffin-embedded and frozen cervical tissue sections (unpublished work). These results suggest that the bDNA ISH method may be an effective means for nucleic acid detection in a variety of specimen types.
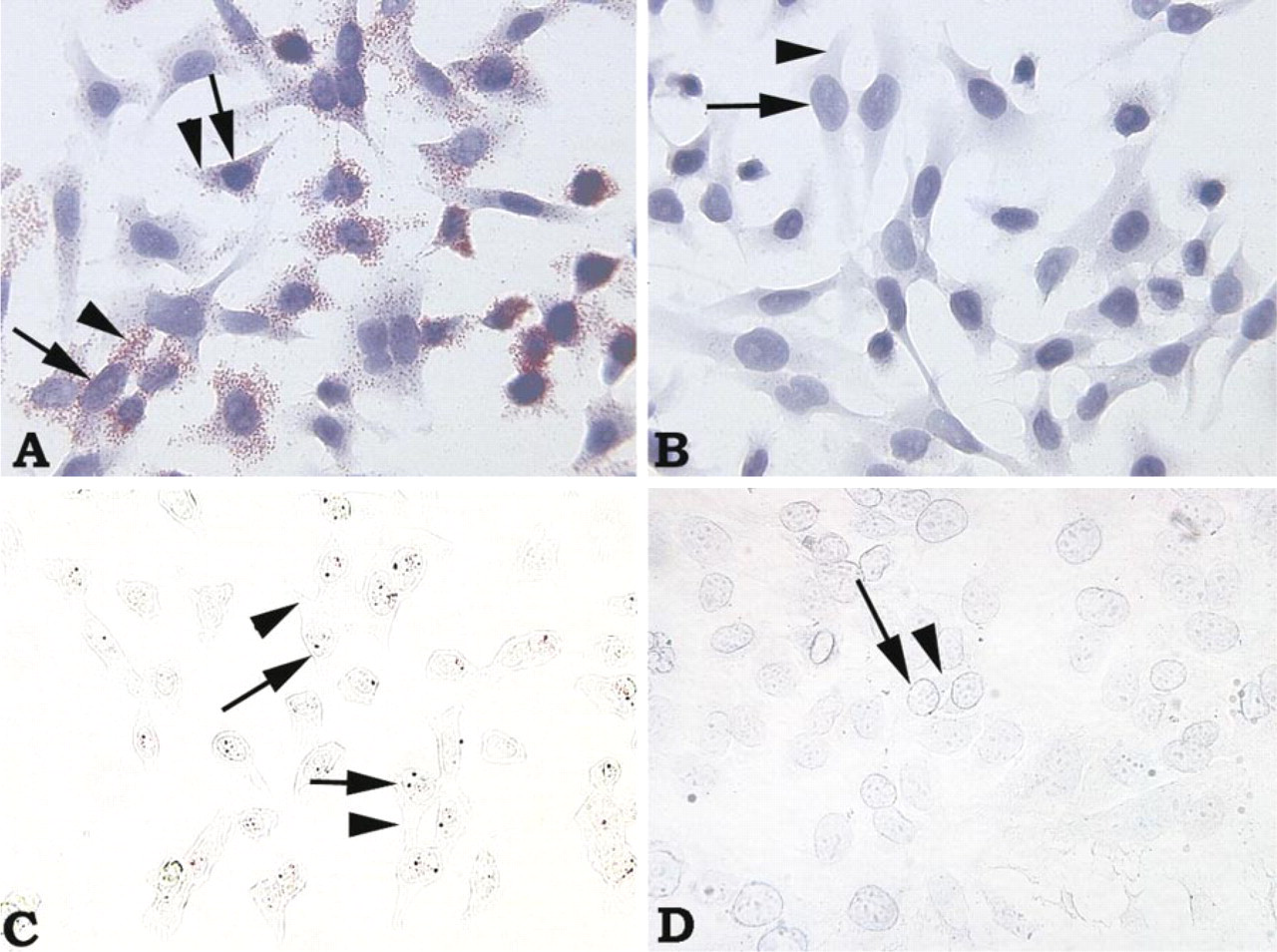
Localization of HPV-18 mRNA predominantly in cytoplasm and HPV-18 DNA in nuclei of HeLa cells by bDNA ISH. (
Another signal amplification system that has been applied to ISH is the catalyzed reporter deposition tyramide signal amplification (CSA) ISH method, which utilizes biotinylated probes, biotinyl tyramide, and streptavidin-conjugated horseradish peroxidase (Schmidt et al. 1997). Our results with the bDNA ISH method agree with other studies that have detected HPV-16 DNA in SiHa cells using CSA (Adler et al. 1997). In our lab, we compared CSA ISH using the GenPoint System (DAKO) with bDNA ISH and found that the two methods have similar sensitivities for detecting HPV-16 DNA in SiHa cells. We found that the HPV-16-specific signal generated with either the bDNA ISH or CSA ISH method was visualized as one or two dots in SiHa cells, and possibly up to four dots in replicating cells. Studies have shown that there is principally one HPV chromosomal integration site in SiHa cells (Baker et al. 1987; Mincheva et al. 1987). It has been suggested that the dots detected probably reflect HPV copies per cell (Siadat–Pajouh et al. 1994).
A couple of features distinguish the bDNA ISH method from CSA ISH. One significant difference is in the probe design. Whereas the HPV-16-specific probes for the bDNA ISH method consist of a set of synthetic oligonucleotides less than 30 bases in length that specifically recognize the E6 and E7 viral genes and transcripts, the probes for the DAKO CSA ISH kit are restriction fragments of a 7-kb biotin-labeled cDNA probe that recognize the E1, E2, E3, E4, E5, E6, and E7 open reading frames (GenPoint System; DAKO Corporation reagent data sheet). The smaller probes generated for bDNA ISH are easier to synthesize and can detect smaller target sequences, thereby providing greater flexibility than longer probes. In principle, the degree of signal amplification can be controlled by adjusting the number of target probes utilized. We have noted that fewer probes are necessary for visualization of HPV in cells with higher HPV copies. Although we did not determine the fewest number of target probes needed for HPV detection with the bDNA ISH method, we found in the course of method development that 15 target probes were sufficient for detection of HPV-16 in SiHa cells (containing one to five HPV copies) and two target probes were sufficient for detection of HPV-16 in CaSki cells (containing 500 HPV copies). Smaller probes also provide more flexibility in obtaining the desired specificity. For example, degenerate target probe sets have been designed for equal detection of several different viral genotypes in the microwell format for HBV, HCV, and HIV (Hendricks et al. 1995; Detmer et al. 1996; Kern et al. 1996). By comparison, as was done for this study, target probe sets can be designed so that detection is genotype-specific. Genotype-specific detection is clinically important for HPV DNA testing because only a subset of HPV genotypes is associated with cervical cancers and high-grade precursor lesions (Manos et al. 1999). Another feature that distinguishes bDNA ISH from CSA ISH is that bDNA ISH does not use an avidin–biotin signal amplification system and hence is not affected by binding of avidin-conjugated reporter molecules to endogenous biotin. By providing greater flexibility in probe design and avoiding potential interference by endogenous biotin, the bDNA ISH method represents a significant advance in low-copy nucleic acid detection.
Compared to PCR-based methods, the bDNA signal amplification system offers a number of advantages for ISH. Because bDNA ISH is based on the sequential hybridization of synthetic DNA probes, it does not require any DNA or RNA polymerase activity and hence is not affected by the presence of polymerase inhibitors in specimens. The potential for diffusion of amplification products away from the target site, which is a concern with PCR-based ISH methods (Nuovo et al. 1991; Wiedorn et al. 1999), also is not a concern with bDNA ISH. Because the AP-conjugated reporter molecule that provides the signal is tethered to the spatially fixed nucleic acid target in the bDNA ISH method, the AP–Fast Red reaction product is retained at the target site. Another advantage is that the bDNA ISH method does not require repeated cycling through elevated temperatures. Because repeated incubation at high temperatures can damage delicate cell morphology, avoiding high-temperature incubations is important for applications in which preservation of intricate cell morphology is important.
In summary, we have developed a bDNA ISH method for sensitive detection of DNA target sequences that overcomes many of the challenges facing ISH techniques today. Based on bDNA signal amplification technology, the bDNA ISH method is highly sensitive (can detect one or two copies of DNA target per cell), specific, and provides subcellular localization of the target sequence. By modifying a few steps in the bDNA ISH procedure, detection of mRNA or DNA targets can be achieved using the same set of DNA oligonucleotide probes. The bDNA ISH method is nonisotopic, rapid (yields results within a day), can be adapted to generate chromogenic and/or fluorescent signals, and should be amenable to automation and quantification. Given its ease of use and reliability, the bDNA ISH method is an attractive alternative for sensitive detection of nucleic acid sequences in a well-preserved morphological context.
Footnotes
Acknowledgements
Acknowledgments
We wish to thank Peter Dailey for helpful discussions about assay design and for review and comments on the manuscript, and Linda Wuestehube for writing and editorial assistance.