
Abstract
Immunohistochemistry is a widely accepted tool to investigate the presence and immunolocalization of cytokines in tissue sections at the protein level. We have tested the specificity and reproducibility of IFNγ immunohistochemistry on tissue sections with a large panel of anti-IFNγ antibodies. Thirteen different commercially available anti-IFNγ antibodies, including seven advertised and/or regularly applied for immunohistochemistry/-cytochemistry, were tested using a three-step streptavidin–biotin–peroxidase technique and a two-step immunofluorescence (FACS) analysis. Immunoenzyme double staining was used to identify the IFNγ-positive cells. Serial cryostat sections were used of human reactive hyperplastic tonsils, rheumatoid synovium, and inflammatory abdominal aortic aneurysms, known to possess a prominent Th1-type immune response. In vitro phorbol myristate acetate/ionomycin-stimulated T-cells served as positive control; unstimulated cells served as negative control. Cultured T-cells were used adhered to glass slides (immunocytochemistry), in suspension (FACS), or snap-frozen and sectioned (immunohistochemistry). Immunocytochemistry and FACS analysis on stimulated cultured T-cells showed positive staining results with 12 of 13 anti-IFNγ antibodies. However, immunohistochemistry of sectioned stimulated T-cells was negative with all. Unstimulated cells were consistently negative. IFNγ immunohistochemical single- and double staining analysis of the tissue sections showed huge variations in staining patterns, including positivity for smooth muscle cells (n = 8), endothelial cells (n = 4), extracellular matrix (n = 4), and CD138+ plasma cells (n = 12). Specific staining of T-cells, as the sole positive staining, was not achieved with any of the 13 antibodies. IFNIFNγ-immunohistochemistry appears unreliable because of lack of specificity to stain T-cells in situ. In fact, depending on the type of anti-IFNγ antibody used, a variety of different cell constituents were nonspecifically stained. Consequently, data based on IFNγ-immunohistochemistry must be interpreted with great caution.
Interferon-gamma (IFNγ) is a 34–50-kD cytokine exclusively produced by T-cells and natural killer cells (Farrar and Schreiber 1993), which plays a key regulatory role in specific immunity. For this reason, IFNγ has been widely investigated in immunological research. In addition to in vitro studies, many investigators also attempted to demonstrate IFNγ protein immunohistochemically in healthy and diseased tissues. For example, studies have been undertaken to localize IFNγ in rheumatoid arthritis (Ulfgren et al. 1995; Smeets et al. 1998a,b), atherosclerosis (Hansson et al. 1989; Frostegard et al. 1999), and inflammatory bowel disease (Camoglio et al. 1998). Cytokine immunohistochemistry is frequently used in combination with molecular biological techniques such as in situ hybridization (ISH) and polymerase chain reaction (RT-PCR) to gain insight into the biological role of cytokines in diseased tissues. However, our own immunohistochemical experiences with several anti-IFNγ antibodies have raised doubts concerning the specificity and the consistency of the staining results obtained. This provides the background for the present study, in which we verified the specificity of anti-IFNγ immunohistochemical staining using a panel of commercially available poly- and monoclonal antibodies.
Materials and Methods
Human Tissue Samples
Tonsils with hyperplastic reactive changes (n = 3, age 3–6 years), inflammatory abdominal aortic aneurysms (IAAA) (n = 2, age 60 and 75 years), and synovial tissue specimens from knee joint with rheumatoid arthritis (RA synovium) (n = 3, age 62–77 years) were obtained at surgery. These tissue samples were used as positive controls for IFNγ staining, based on the expression of IFNγ mRNA as detected with RT-PCR (Ramshaw et al. 1994; Harabuchi et al. 1996; Kotake et al. 1997). Moreover, IFNγ protein was detected with immunohistochemistry in tonsil and RA synovium (Hoefakker et al. 1993; Andersson et al. 1994; Dolhain et al. 1996; Smeets et al. 1998a,b) and ELISA in IAAA (Szekanecz et al. 1994). Non-atherosclerotic vessel segments (n = 4, age 10–14 years) were used as controls. All tissue samples were snap-frozen in liquid nitrogen and stored at –80C. Six-μm cryostat serial sections were cut, and stored at –80C.
Cell Specimens
Cultured T-cell lines from human aortic atherosclerotic plaques were prepared as previously described (De Boer et al. 1999). Cells were cultured (5 × 106 /ml) in Iscoves modification of Dulbecco's medium (IMDM; Gibco BRL Life Technologies, Paisley, Scotland) supplemented with 10% pooled human serum in the presence or absence of 10 ng/ml phorbol-12-myristate-13-acetate (PMA) (Sigma; St Louis, MO) and 100 ng ionomycin (Calbiochem; La Jolla, CA) in 25-cm2 culture flasks. Monensin (Sigma) was added (2.88 μM/ml in dimethylsulfoxide) to prevent active cytokine excretion, thus resulting in intracellular accumulation of IFNγ. After 4 h, the cell suspensions were washed three times with PBS without protein additives and divided into three fractions. The first fraction was adhered overnight at 4C on Bio-Rad adhesion slides (Hercules, CA), 2 × 104 cells per spot. After brief washing with PBS, these cell specimens were stored in PBS at 4C and used for immunocytochemistry within 2 days. The second fraction was concentrated in an Eppendorf tube in 100 μl PBS (2 × 105 cells), mixed gently with 100 μl Tissue-Tek OCT compound (Sakura; Zoeterwoude, The Netherlands), and snap-frozen in liquid nitrogen. Six-μm cryostat sections were cut and handled as described for tissue cryostat sections. The remaining cells were used for FACS analysis.
Immunostaining Reagents
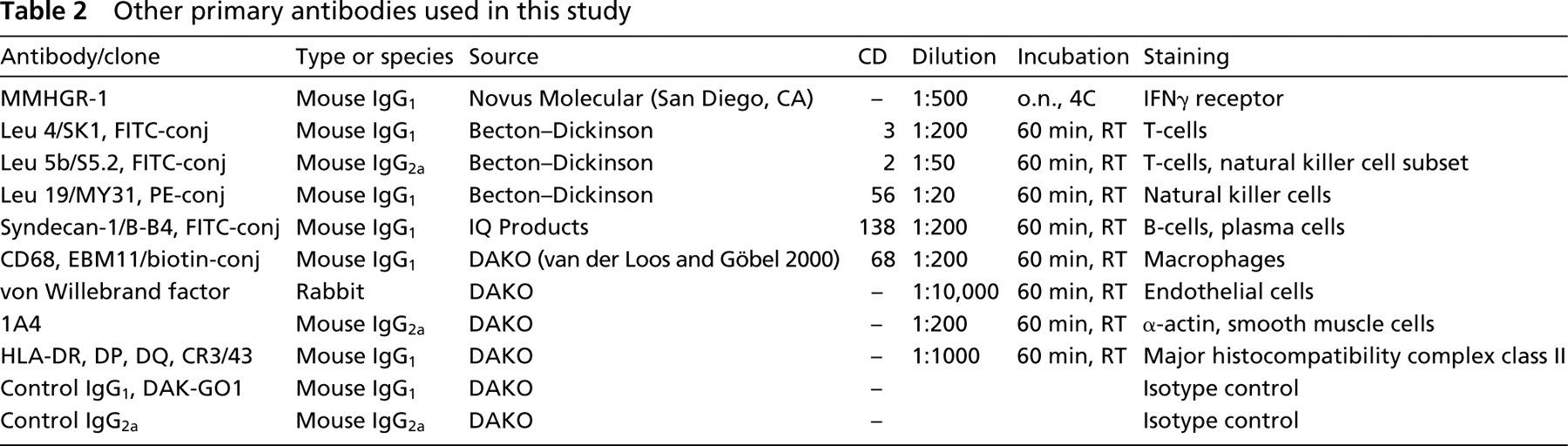
We employed 13 anti-human anti-IFNγ poly- and monoclonal antibodies, as listed in Table 1. As indicated in Table 1, seven anti-IFNγ antibodies were advertised and/or regularly applied for immunocytochemistry/-histochemistry. Other primary antibodies used in this study are listed in Table 2.
Biotinylated goat anti-mouse Ig (GAM/bio), biotinylated goat anti-rabbit Ig (GAR/bio), alkaline phosphatase-conjugated goat anti-rabbit Ig (GAR/AP), normal mouse serum (NMS), normal goat serum (NGS), normal swine serum (NSS), AP-conjugated streptavidin (streptavidin/AP), streptavidin–biotin complex with HRP (SABC/HRP), rabbit antifluorescein (rabbit anti-FITC), and endogenous biotin blocking kit were from DAKO (Glostrup, Denmark). Phycoerythrin (PE)-conjugated GAM, PE-conjugated GAR, biotinylated goat anti-mouse IgG2a (GAM-IgG2a/bio), and HRP-conjugated goat anti-mouse IgG1 (GAM-IgG1/HRP) were from Southern Biotechnology Associates (Birmingham, AL). Biotinylated and PE-conjugated swine anti-goat Ig (SAG) were from BioSource (Nivelles, Belgium). Rabbit anti-phycoerythrin (rabbit anti-PE) was from Biogenesis (Poole, UK). β-Galactosidase-conjugated streptavidin (streptavidin/GAL) was from Boehringer/Roche (Mannheim, Germany). PowerVision-AP-conjugated goat anti-rabbit Ig (PowerVision-GAR/AP) was from ImmunoVision Technologies (Daly City, CA). 3-Amino-9-ethylcarbazole (AEC), naphthol-AS-MX-phosphate, Fast Blue BB (cat. no. 3378), and saponin (cat. no. 7900) were from Sigma.
Anti-human IFNγ antibodies used in this study
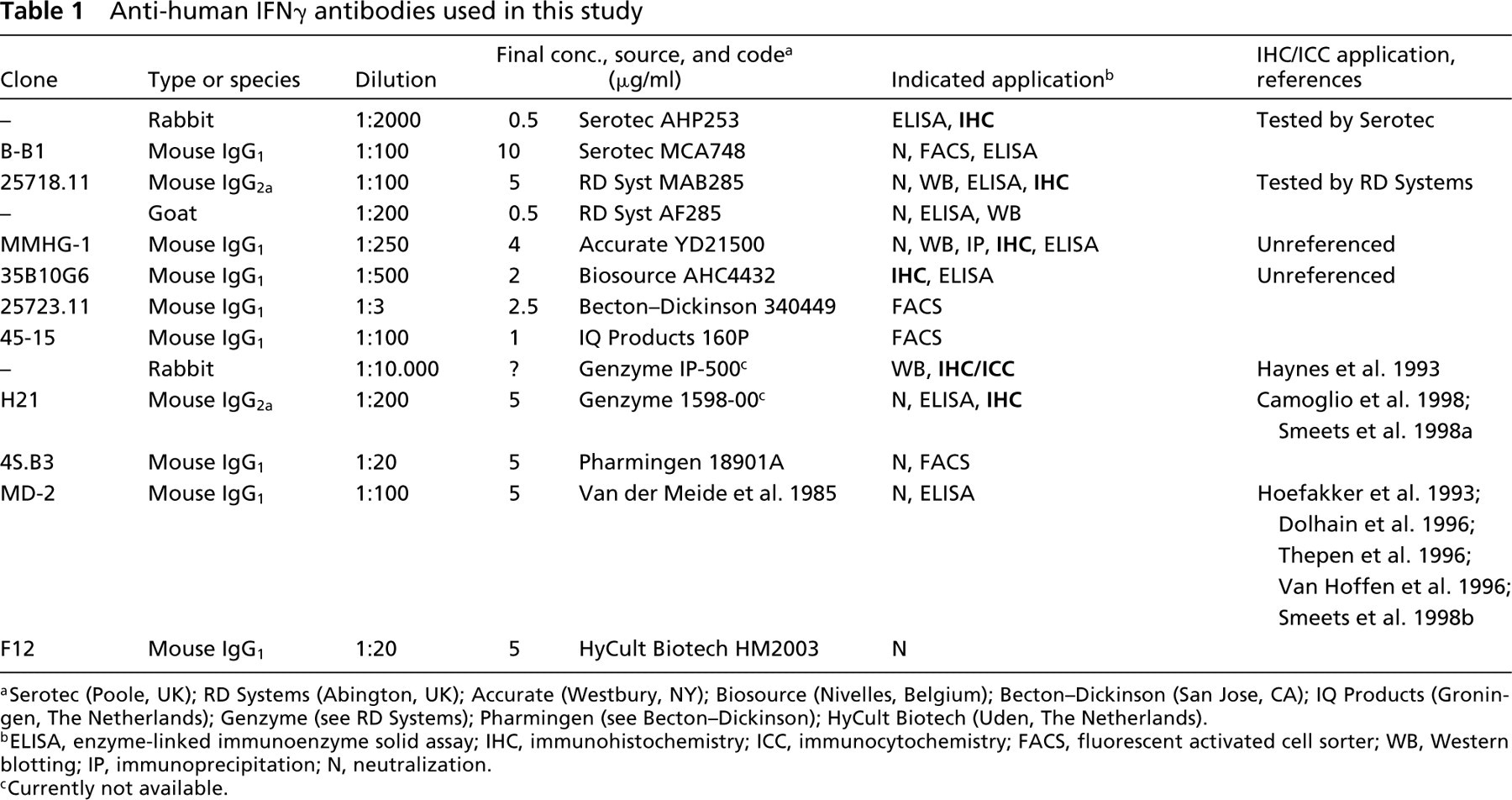
aSerotec (Poole, UK); RD Systems (Abington, UK); Accurate (Westbury, NY); Biosource (Nivelles, Belgium); Becton–Dickinson (San Jose, CA); IQ Products (Groningen, The Netherlands); Genzyme (see RD Systems); Pharmingen (see Becton–Dickinson); HyCult Biotech (Uden, The Netherlands).
bELISA, enzyme-linked immunoenzyme solid assay; IHC, immunohistochemistry; ICC, immunocytochemistry; FACS, fluorescent activated cell sorter; WB, Western blotting; IP, immunoprecipitation; N, neutralization.
cCurrently not available.
Other primary antibodies used in this study
Immunocytochemistry and Immunohistochemistry
Cell specimens and cryostat tissue sections were fixed in either acetone (10 min, 4C) or 4% paraformaldehyde (PFA) in PBS (5 min, room temperature). When PFA fixation was used, 0.1% saponin was added for membrane permeabilization (Sander et al. 1991; Dolhain et al. 1993) to all incubation steps. Endogenous peroxidase activity was blocked with 0.1% sodium azide + 0.3% peroxide in 50 mM Tris-HCl-buffered saline, pH 7.8 (TBS) (20 min, RT) (Li et al. 1987). Endogenous biotin was blocked with subsequent incubations of 0.1% avidin and 0.01% d-biotin from the DAKO endogenous biotin blocking kit (twice for 15 min at RT). Primary antibodies and antibody-enzyme conjugates were diluted in TBS + 1% bovine serum albumin (BSA). Single immunostaining consisted of overnight incubation at 4C with the selected primary anti-IFNγ antibodies (Table 1) and standard SABC/HRP detection. HRP activity was visualized with AEC (0.5 mg/ml) and peroxide (0.01%) in acetate buffer (pH 5.2, 50 mM).
Controls consisted of replacing the primary antibody with a non-immune mouse antibody of identical subclass (DAKO). Specific IgG concentration and Ig isotype/subclass were matched with the specific primary antibody from the original single-staining experiment.
Immunoenzyme Multiple Staining of Tissue Sections
A double-staining procedure based on a multistep technique (van der Loos 1999) was applied for comparison of IFNγ immunostaining with different cellular markers. The staining procedure consisted of the following subsequent incubation steps: NGS (1:10, 15 min), IFNγ antibody (clones MMHG-1 1:100, MD-2 1:50 or rabbit antibody Genzyme IP-500 1:5000, all overnight at 4C), GAM/bio (1:200, 30 min) or GAR/bio (1:400, 30 min), SABC/HRP (1:100, 30 min), and NMS (1:10, 15 min). For the goat anti-IFNγ antibody, the first part of the double staining consisted of NSS (1:10, 15 min), IFNγ goat antibody (1:100, overnight at 4C), SAG/biotin (1:100, 30 min), SABC/HRP (1:100, 30 min), and NGS (1:10, 15 min). Then, a panel of directly conjugated primary antibodies was applied: FITC-conjugated anti-CD3, CD2, CD138, biotinylated anti-CD68 (van der Loos and Göbel 2000), and PE-conjugated CD56 (60 min) (Table 2). For the FITC- and PE-conjugated antibodies, further steps were as follows: rabbit anti-FITC (1:1000, 15 min), or rabbit anti-PE (1:200, 15 min) and PowerVision-GAR/AP (undiluted, 30 min). The biotinylated antibody was followed by streptavidin/AP (1:100, 30 min). The enzymatic activities of AP and HRP were visualized in blue (Fast Blue BB) and red (AEC), respectively (van der Loos 1999).
Two triple immunoenzyme stainings using β-galactosidase (turquoise), AP (red), and HRP (brown) as marker enzymes, and blue nuclear counterstain (van der Loos 1999), were performed to monitor the major cell types present in the tissue specimens. The first consisted of anti-α-actin (mouse IgG2a) and anti-CD68 (mouse IgG1), combined with FITC-conjugated anti-CD3, in a multistep staining procedure. Anti-α-actin and anti-CD68 were incubated in a cocktail, followed by GAM-IgG2a/biotin (1:50), GAM-IgG1/HRP (1:50) in a cocktail (30 min) and streptavidin/GAL (1:40, 30 min). After a blocking step with normal mouse serum (1:10, 15 min), incubation was performed with FITC-conjugated anti-CD3, rabbit anti-FITC (1:1000, 15 min), and GAR/AP (1:20, 30 min). Smooth muscle cells, macrophages, and T-cells were immunostained in turquoise, brown, and red, respectively. The second consisted of anti-α-actin (mouse IgG2a), anti-CD68 (mouse IgG1), and anti-von Willebrand factor (rabbit) in one cocktail, followed by GAM-IgG2a/bio (1:50), GAM-IgG1/HRP (1:50), GAR/AP (1:20) in cocktail (30 min), and streptavidin/GAL (1:40, 30 min). Smooth muscle cells, macrophages, and endothelial cells were immunostained in turquoise, brown, and red, respectively.
FACS Analysis
All 13 anti-IFNγ antibodies were applied for indirect intracellular FACS staining. In vitro PMA/ionomycin-stimulated T-cells were fixed with 1% PFA in PBS (10 min, 4C) and treated with 0.1% saponin for permeabilization (Sander et al. 1991). Final concentrations of the primary antibodies diluted in PBS with 0.1% saponin and 1% BSA (60 min, 4C) were similar as mentioned for immunohistochemistry/cytochemistry in Table 1. Depending on the primary antibody, PE-conjugated GAM, GAR, or SAG (all 1:200) was used as second-step reagent (30 min, 4C). Fluorescence of 104 cells was analyzed on a FACS Calibur (Becton Dickinson, Richmond, CA).
Leakage of IFNγ from Tissue Sections
From the frozen stimulated and unstimulated cultured T-cells, 20 6-μm sections were cut and mounted on organosilanecoated slides. Either unfixed, acetone-fixed (10 min, 4C, air-dried) or PFA-fixed (5 min, RT, briefly washed three times with TBS) sections were encircled with a wax pen and covered with 100 μl TBS for 30 min. Fifty-μl samples were taken from the buffer covering the tissue sections and subjected to IFNγ ELISA. A Pelikine IFNγ kit was used, following the instructions as supplied by the manufacturer (CLB; Amsterdam, The Netherlands).
Results
IFNγ Staining Characteristics with Cultured T-cells
Stimulated intact T-cells on adhesion slides showed a distinct signal in the majority of the cells with 12 of 13 anti-IFNγ antibodies (Figure 3E). Unstimulated cells were negative or occasionally weakly positive (Figure 3B). Stimulated T-cells showed a much stronger staining intensity after PFA fixation (Figure 3E) than after acetone fixation (Figure 3D). These immunocytochemical staining results were completely confirmed by FACS analysis (Table 3).
An interesting observation was that IFNγ immunohistochemistry was consistently negative after cryosectioning of stimulated T-cells, (Table 3; Figure 3F), even after raising the primary antibody concentration to 50 μg/ml. This result was found after both acetone and PFA fixation.
To test the possible leakage of IFNγ from either unfixed, acetone-, or PFA-fixed cultured T-cell sections, buffer covering the specimens was subjected to ELISA. Buffers covering unfixed sections from stimulated T-cells contained IFNγ protein at 14.4–49.3 pg/ml (n = 4) and concentrations below detection limit (± 10 pg/ml) for the unstimulated cells. After acetone or PFA fixation, only values close to or below detection limit were found. Performance of this leakage test with the other tissue specimens failed because IFNγ values with unfixed sections were too low.
IFNγ Staining Characteristics with Tissue Sections
Comparison of the IFNγ single staining with triple immunostainings marking the major cellular components in tonsil, RA synovium, and IAAA indicated positive IFNγ staining of various tissue components with a preference for smooth muscle cells (n = 8), endothelial cells (n = 4), extracellular matrix (n = 4), macrophages (n = 1), and nerve bundles (n = 2). Considering only those IFNγ antibodies that were advertised and/or regularly applied for immunohistochemistry (n = 7; Table 1), there was no consistent staining pattern either. The positive cell types included smooth muscle cells (n = 4), endothelial cells (n = 3), extracellular matrix (n = 2), macrophages (n = 1), and nerve bundles (n = 1). Moreover, distinct expression of IFNγ with T-cells was not observed. Remarkably, a strong IFNγ smooth muscle cell positivity was also found in the media of non-atherosclerotic and non-inflamed aortic segments (Figure 1K). Apart from these various cellular constituents a distinct, consistently positive cell population was observed with 12 out of 13 antibodies. This characteristic cell population was found exclusively positive without additional staining of other cellular elements with three anti-IFNγ antibodies: clones MMHG-1, 35B10G6, and RD Systems goat antibody (Table 3). These positive cells were found in relatively low amounts in tonsil and IAAA but were abundant in the RA synovium (Figures 2B-2D). Negative control experiments did not show any staining (Figures 1I and 2A). Typical results of anti-IFNγ immunohistochemistry of all three tissues are summarized in Table 3 and illustrated in Figures 1A-1H and Figures 2B-2D.
Anti-human IFNγ antibodies applied in FACS, ICC, and IHC a
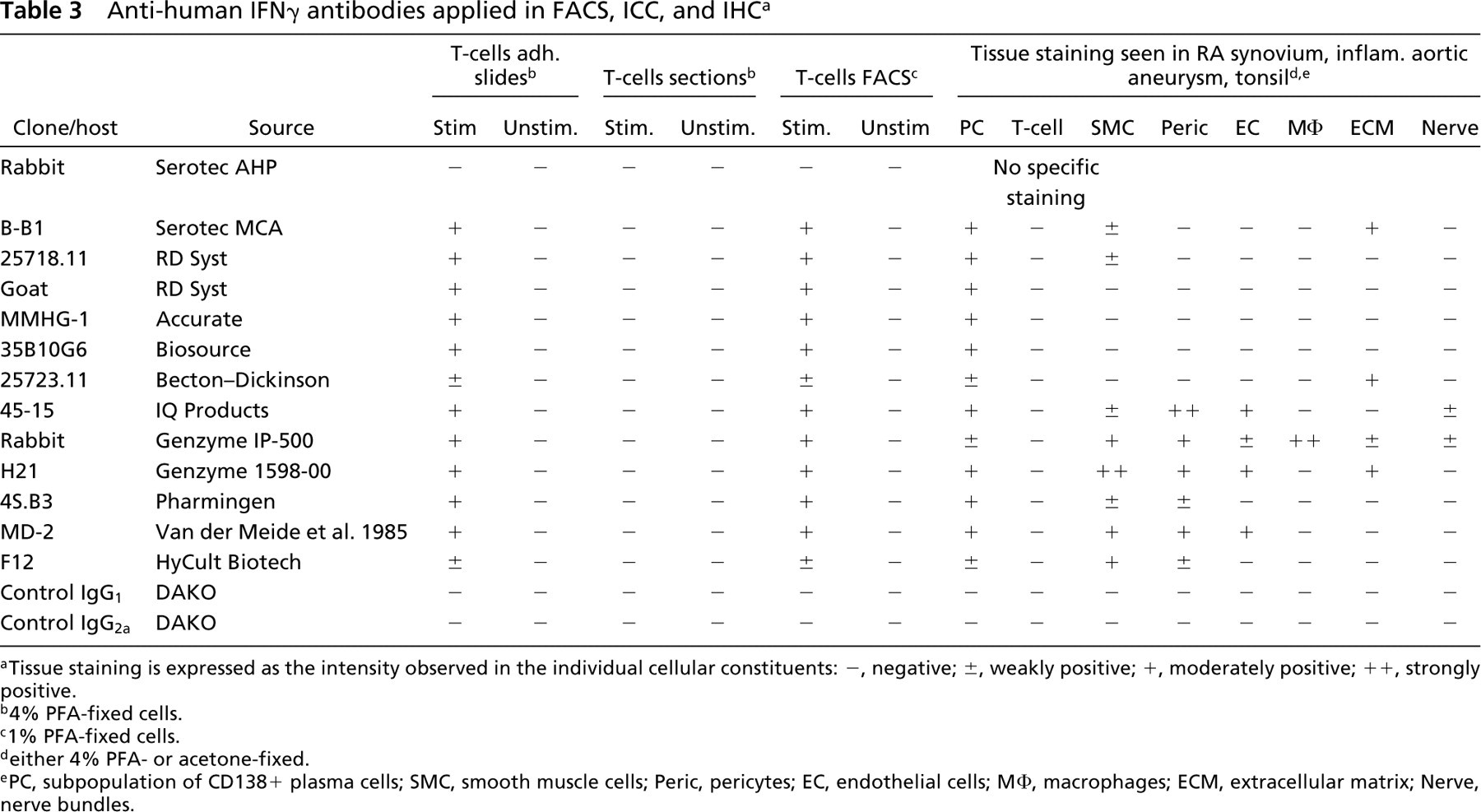
aTissue staining is expressed as the intensity observed in the individual cellular constituents: –, negative; ±, weakly positive; +, moderately positive; ++, strongly positive.
b4% PFA-fixed cells.
c1% PFA-fixed cells.
deither 4% PFA- or acetone-fixed.
ePC, subpopulation of CD138+ plasma cells; SMC, smooth muscle cells; Peric, pericytes; EC, endothelial cells; MΦ, macrophages; ECM, extracellular matrix; Nerve, nerve bundles.
Comparison of acetone and PFA as fixatives before IFNγ immunostaining showed similar results with respect to localization and staining intensity. Raising the antibody concentration (clones MMHG-1, MD-2, Genzyme rabbit antibody, and RD Systems goat antibody) up to 50 μg/ml, revealed over-stained images with intensely positive cells as described above.
To investigate the possible binding of IFNγ to its concomitant receptor, tissue localization of IFNγ immunostaining was compared with IFNγ receptor staining in serial sections. In RA synovium, IAAA, and tonsil, the anti-IFNγ receptor antibody showed distinct positive staining of endothelial cells, macrophages, and weak staining of smooth muscle cells (Figure 1J), which could only partly be co-localized with the anti-IFNγ staining (Figures 1B-1H). Moreover, medial smooth muscle cells in non-atherosclerotic aortas were IFNγ receptor (Figure 1L) -negative, which contrasted with the strong IFNγ positivity (Figure 1K).
Cellular Specificity of Anti-IFNγ Antibodies
To reveal the characteristic cell type showing positive staining with almost all anti-IFNγ antibodies, clones MMHG-1, MD-2, Genzyme rabbit antibody, and RD Systems goat antibody were subjected to immunoenzyme double-staining experiments with cellular markers on the RA synovium and tonsil cryostat sections. It appeared that T-cells (CD2, CD3), natural killer cells (CD56), and macrophages (CD68) did not co-localize with the IFNγ-positive cell population (Table 3). However, a subpopulation of approximately 5% of all CD138 positive plasma cells co-localized with IFNγ-positive cells, observed as a purple mixed-color in the cytoplasm (Figures 4A-4D).
Discussion
In the present study we have compared the immunohistochemical applicability of 13 different anti-IFNγ antibodies on cryostat tissue sections. IFNγ immunohistochemical single- and double-staining analysis of the tissue sections showed a large variation in staining patterns, but positive staining of T-lymphocytes was never observed. In this context, it should be emphasized that not all 13 IFNγ-antibodies used in this study were originally developed and tested for an immunohistochemical application. This also could explain the highly variable immunohistochemical staining patterns (Figures 1A-1H), despite the fact that the antibodies were capable of detecting IFNγ in cultured and in vitro-stimulated intact T-cells (Table 3). For example, anti-IFNγ clone MD-2 was first described for ELISA application by Van der Meide et al. (1985) and only much later was applied for immunohistochemical purposes (Hoefakker et al. 1993). The latter application is subsequently cited by other investigators and has led to the use of this antibody on tissue sections (Dolhain et al. 1996; Thepen et al. 1996; Van Hoffen et al. 1996; Smeets et al. 1998b).
We also verified the immunoreactivity of the anti- IFNγ antibodies on intact PMA/ionomycin-stimulated T-cells, known to contain substantial amounts of IFNγ. Intact in vitro-stimulated T-cells showed a typical intracellular IFNγ staining pattern (Figure 3E) similar to the findings by others (Sander et al. 1991; Dolhain et al. 1993; Andersson and Andersson 1994; Krouwels et al. 1997). In addition, 12 of 13 anti-IFNγ antibodies subjected to FACS analysis showed positive staining with in vitro-stimulated T-cells, whereas unstimulated cells were negative. These experiments reveal that 12 of 13 anti-IFNγ antibodies have the potential to recognize IFNγ in in vitro-stimulated intact T-cells. In contrast to these intact in vitro-stimulated T-cells, IFNγ immunohistochemistry performed on cryosections prepared from these cells gave negative results (Figure 3F). Given the fact that these in vitro PMA/ionomycin-stimulated T-cells contain higher levels of IFNγ protein than activated T-cells in vivo (Del Prete et al. 1991), it may be not surprising that IFNγ immunostaining of T-cells in tissue sections of IAAA, RA synovium, and tonsil was negative.
The present experiments show that there is a clear discrepancy between the results obtained with cultured intact T-cells and those with cryosectioned T-cells. Apparently, immunocytochemical staining of intact T-cells is not a reliable model system for testing the immunohistochemical applicability of IFNγ antibodies on cryostat tissue sections, as has been suggested in the literature (Andersson et al. 1994; Dolhain et al. 1996).
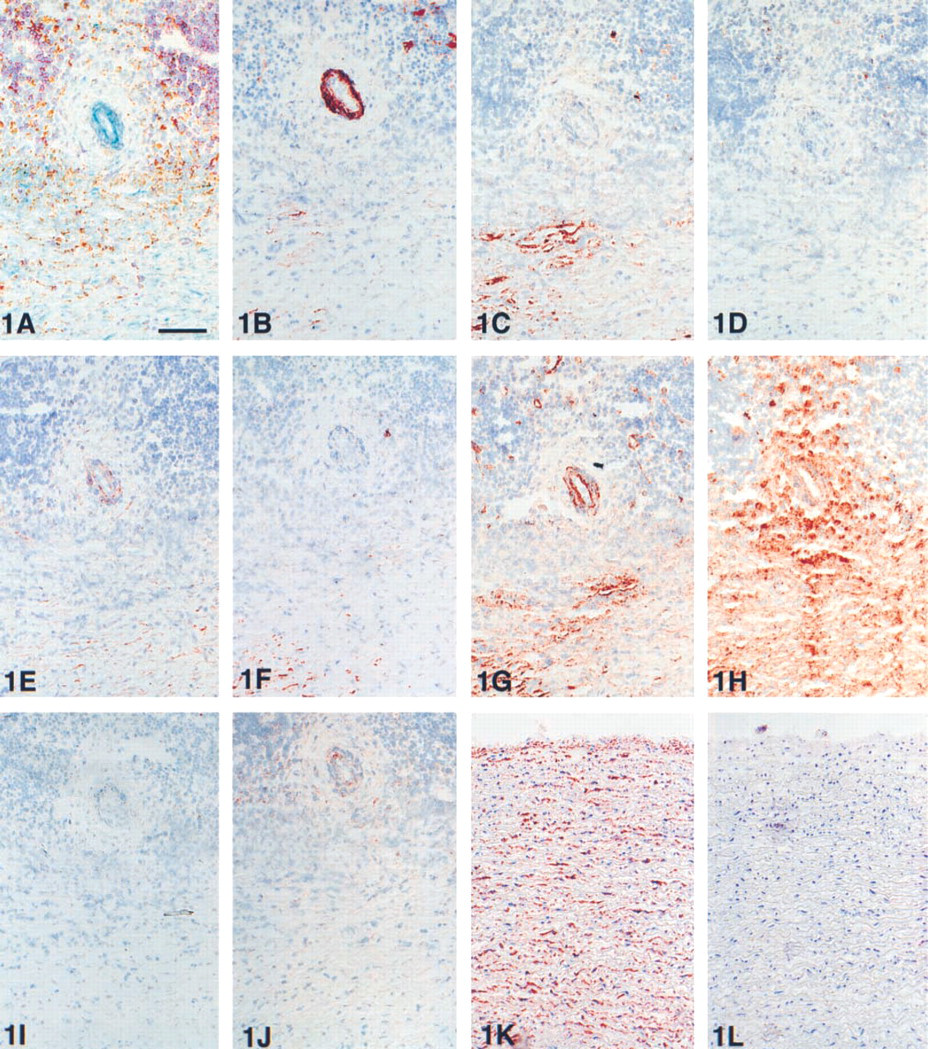
Acetone-fixed cryostat serial sections from an inflammatory abdominal aorta aneurysm segment, showing a detail of the adventitia. A muscular vessel is surrounded by inflammatory cells and fibrotic tissue. (
It is remarkable that in the majority of papers the observed IFNγ positivity simply was assumed to represent T-cells. Confirmation by double staining was performed only by Dolhain et al. (1996), who showed CD3/IFNγ co-localization in RA synovium samples. In this study we could not reproduce those results, even in the same type of tissue. Attempting to demonstrate the co-localization of IFNγ in T-cells, we performed double staining with both CD2 and CD3. Double staining with CD2 was also performed because CD2 is not downregulated after T-cell activation (Moingeon et al. 1991), in contrast to CD3 (Van Lier et al. 1987). Furthermore, we have been able to detect IFNγ/CD3 co-localization in in vitro-stimulated T-cells (not shown). We have no explanation for the different results in our study compared to that of Dolhain et al. (1996).
The obvious difference between in vitro-stimulated intact T-cells and cryostat tissue sections prepared from these cells is the presence of an outer membrane. Therefore, it appears attractive to hypothesize that intact T-cells retain IFNγ more effectively during the fixation procedure, either with acetone or PFA, than a sectioned cell. Possibly IFNγ protein is lost from the cryosectioned T-cells during the fixation procedure due to extraction of the antigen (Larsson 1993) or due to poor fixation. Arguments in favor of this leakage-hypothesis are the observations that (a) IFNγ is detectable by ELISA in the buffer covering the unfixed in vitro-stimulated T-cell cryostat sections. However, after fixation of the T-cell cryostat sections with either acetone or PFA, almost no IFNγ is detectable in the overlying buffer, whereas the immunohistochemical visualization of IFNγ in cryosectioned stimulated T-cells was completely negative also (Figure 3F). (b) Acetone fixation of intact T-cells, which completely dissolves the fatty membrane structures, shows a near-negative IFNγ staining result (Figure 3D), whereas the same cells were intensely positive after PFA fixation (Figure 3E).
IFNγ immunohistochemistry on cryostat tissue sections from tonsil, RA synovium, and IAAA revealed highly variable staining patterns with different antibodies. A variety of cellular constituents were positive, with a high preference for smooth muscle cells. Apart from staining of various cellular constituents, 12 of 13 anti-IFNγ antibodies revealed staining of a CD138-positive plasma cell subset (Figure 4) but not of T-cells. Non-T-cell positivity with anti-IFNγ antibodies has been reported previously and is considered a result of receptor-bound IFNγ (Dolhain et al. 1996). Indeed, some of the IFNγ staining patterns resemble the IFNγ receptor staining pattern (Figure 1J). However, there are indirect arguments that certainly not all non-T-cell IFNγ positivity can be explained as receptor bound. (a) In the non-atherosclerotic aortas, without any morphological signs of inflammation, the medial smooth muscle cells showed massive IFNγ positivity, whereas the IFNγ receptor was negative (Figures 1L and 1K). (b) When non-T-cell IFNγ staining in non-atherosclerotic aortas was caused indeed by receptor-bound IFNγ, this would have induced major histocompatibility complex Class II expression (HLA-DR, DP, DQ). However, this was not observed. This absence of HLA-DR, DP, and DQ positivity in non-atherosclerotic aortas also excludes the possibility that IFNγ receptor staining is perhaps missed because IFNγ blocks the antibody binding site.
Considering the IFNγ positivity of plasma cells, some investigators claimed expression of trace amounts of IFNγ by B-lymphocytes under in vitro conditions (Pang et al. 1992; Jelinek and Braaten 1995). In our opinion, these findings cannot be matched with the abundant and strong IFNγ staining of the plasma cell subpopulation as observed in all three tissues studied. Therefore, apart from the non-T-cell staining of various cellular constituents, also the plasma cell IFNγ-positivity was considered as a staining artifact. This artifactual staining might be caused by the formation of conditional epitopes (Willingham 1999) or redistribution of small molecular weight antigens (Larsson 1993), both of which may occur during the fixation procedure.
The nonspecific positive staining of the plasma cell subpopulation was shown not to be unique for anti- IFNγ antibodies. Using, for example, anti-IL-2 or anti-IL-4 antibodies, known to be secreted by T-cells upon activation, we also observed a strong positivity in RA synovium of the same plasma cell population that was found positive with anti-IFNγ antibodies, whereas positive T-cells were not observed. Non-T-cell IFNγ staining may be the basis for conflicting immunohis tochemical staining results when different anti-IFNγ antibodies were applied to similar tissue specimens. For example, Andersson et al. (1994) reported in chronic recurrent tonsillitis a few IFNγ positive cells using clone DIK-1, whereas Hoefakker et al. (1993) reported massive IFNγ positivity with clone MD-2 in the same tissue. Moreover, the application of extremely high concentrations of IFNγ antibody (MD-2, 45–100 μg/ml) (Thepen et al. 1996; Van Hoffen et al. 1996) for immunostaining of cryostat sections may introduce staining artifacts. Not only may the IFNγ antibodies themselves may play a role but also the application of different tissue fixatives before the IFNγ staining procedure may have an effect on the final staining result. For example, acetone fixation was employed by Hoefakker et al. (1993), Dolhain et al. (1996), Smeets et al. (1998b), and Thepen et al. (1996), whereas PFA fixation and saponin treatment were used by Ulfgren et al. (1995), Smeets et al. (1998a), and Andersson et al. (1994). In our hands, a comparison of these two popular fixatives showed a dramatic loss of IFNγ staining after acetone fixation of intact T-cells (Figures 3D and 3E), confirming the use of PFA fixation as indicated by Andersson et al. (1994). However, in considering the non-T-cell IFNγ staining of tissue sections after either acetone or PFA fixation, there was no difference regarding localization and intensity. This observation is another indication that the non-T-cell staining in tissue sections after acetone fixation has a nonspecific basis. The present description of nonspecific IFNγ immunohistochemical staining shows analogy with the false-positive staining of anti-RAP-5 antibody detecting the ras oncogene product p21 as reported by Samowitz et al. (1988) and Gutheil et al. (1989). Although these investigators employed immunoprecipitation and Western blotting as contra-evidence, they finally proved that the initial RAP-5 immunohistochemical positivity could be recognized as completely nonspecific. Furthermore, Kaino et al. (2000) described the crossreactivity of anti-rat CD45RA antibody with glucagon-producing islet α-cells in formalin-fixed and paraffin-embedded rat pancreatic tissue.
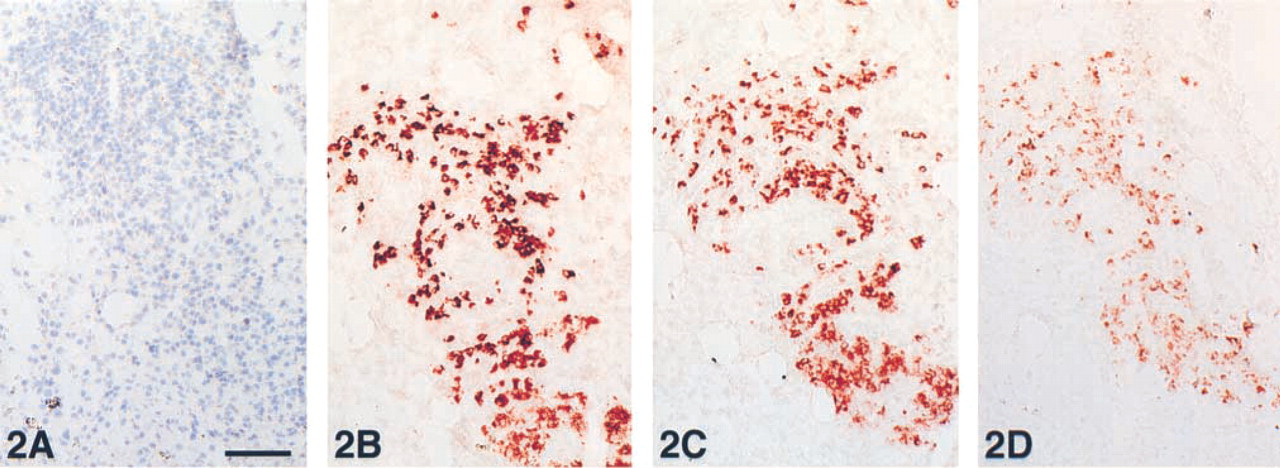
Acetone-fixed cryostat serial sections from a synovium of a rheumatoid arthritis patient. (
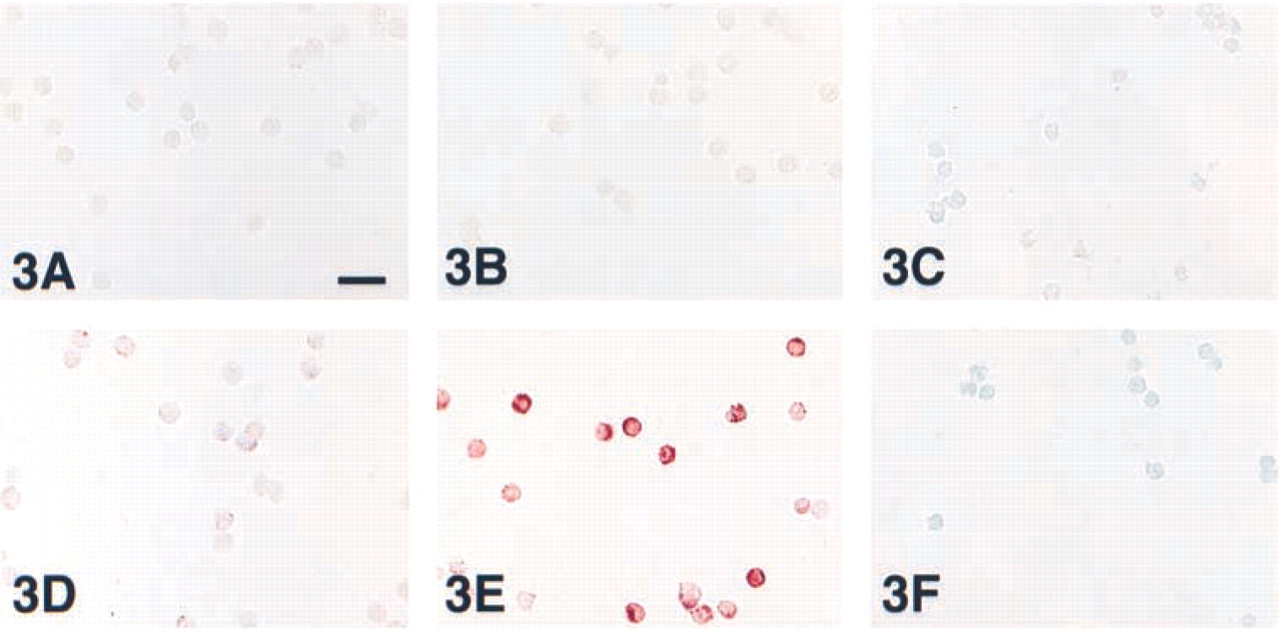
PMA/ionomycin-stimulated (
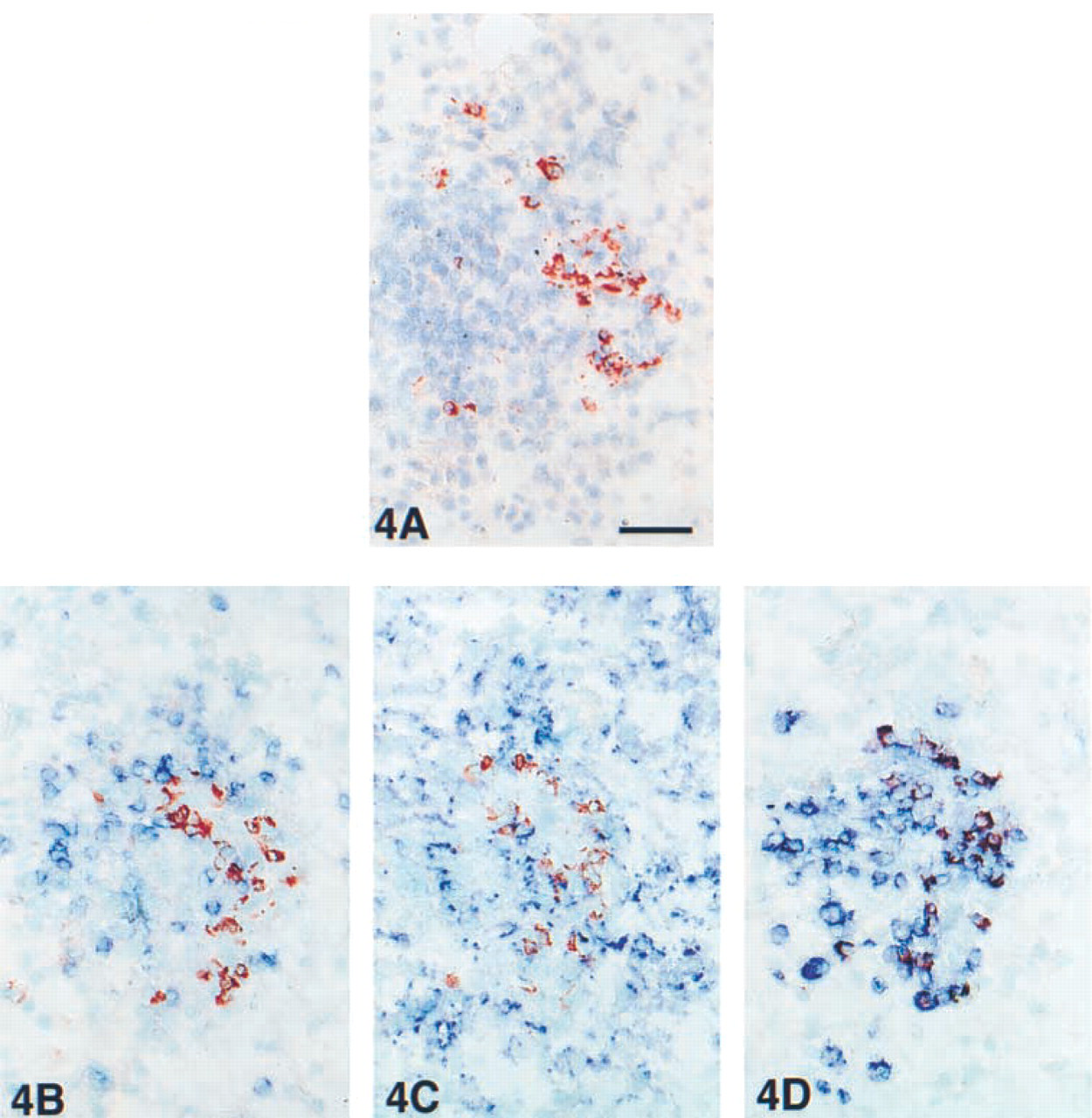
Detail of a synovium with rheumatoid arthritis in four adjacent acetone-fixed cryostat sections, showing a focal area with inflammatory cells. (
PMA/ionomycin in vitro-stimulated (
We consider further testing of the anti-IFNγ antibody specificity using absorption with IFNγ antigen not to be useful. In our opinion and that of others (Burry 2000), an absorption control does not prove the specificity of an antibody for its antigen in a fixed tissue section. For example, anti-IFNγ clone MD-2 was subjected successfully to such an absorption experiment (Hoefakker et al. 1993), but our present data clearly show this antibody as not to be specific for tissue IFNγ staining (Table 3; Figure 1L).
Future Perspectives for IFNγ Immunostaining?
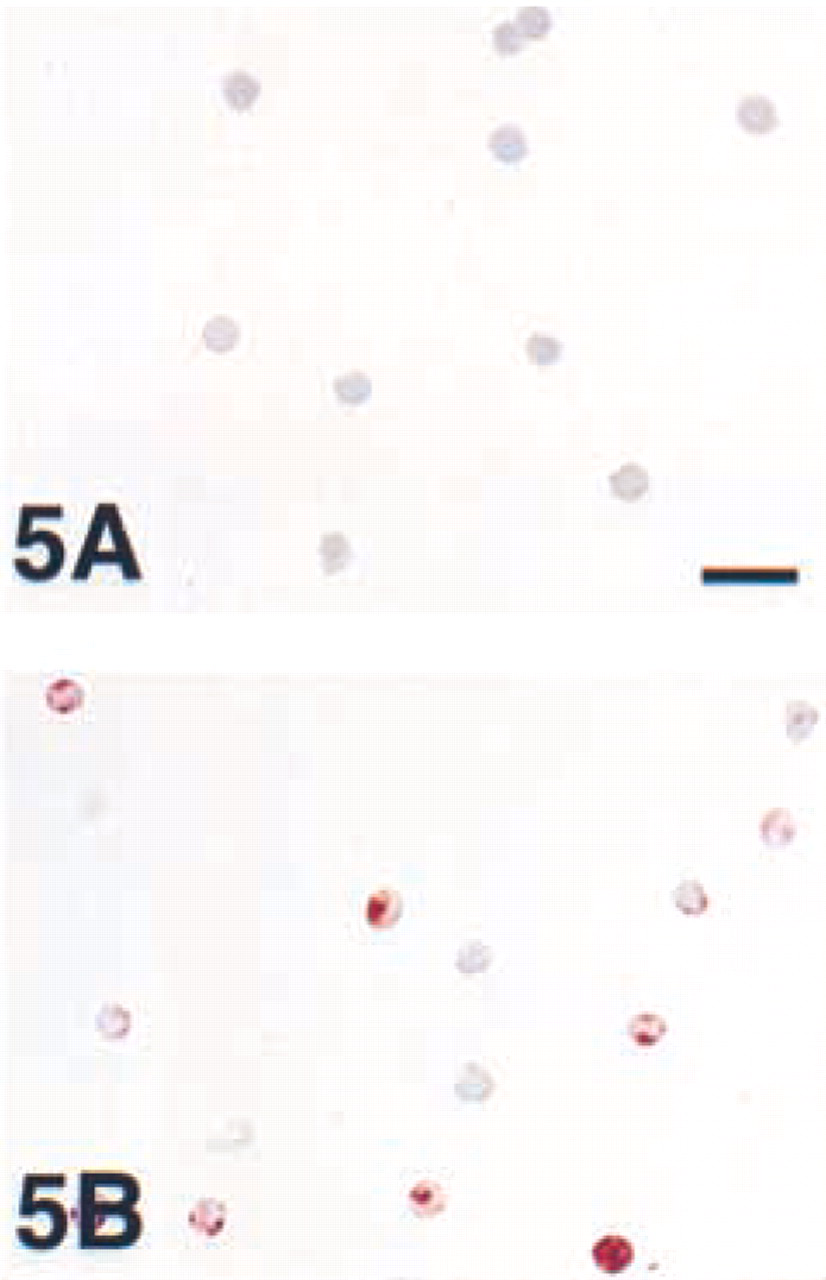
At the request of one of the reviewers and after discussions at the latest Congress of Histochemistry and Cytochemistry, we tested the option of prefixation by perfusion or immersion in PFA before freezing. This method has been applied, for example, by Van Noorden and Polak (1985), Van Noorden (1986), and Larsson (1988), and is especially recommended for detection of small peptides and hormones. However, among the vast majority of reports applying acetone or PFA post-fixation of cryosections, we could find only one report using prefixation for the detection of IFNγ (Kakazu et al. 1999). Our pilot experiment showed that PFA prefixation of in vitro-stimulated T-cells indeed retains and preserves IFNγ after freezing and sectioning (Figures 5A and 5B), in contrast to PFA postfixation (Figure 3F). Apparently, PFA postfixation of a cryosection is too slow (Fox et al. 1985) and cannot prevent leaking of IFNγ from cryosections. However, PFA prefixation of a closed cell allows a firm fixation of IFNγ, thus preventing leakage even from tissue sections. Thus far, however, we have not been able to demonstrate IFNγ immunohistochemically in a few PFA-prefixed and 20% sucrose freeze-protected tissue blocks tested. Nevertheless, it could be that PFA prefixation is the key to successful immunohistochemical visualization of IFNγ and perhaps of other cytokines. Moreover, we suspect that missing the IFNγ staining of activated T-cells in cryosections from PFA prefixed tissue blocks, in contrast to prefixed in vitro-stimulated T-cells, may be a sensitivity problem of current detection systems even when tyramide amplification is used.
Conclusion
Using the present panel of anti-IFNγ antibodies, specific staining of activated T-cells (Th-1 type) in tissue sections is not observed with generally applied staining procedures, a phenomenon most likely due to either the lack of sensitivity and/or to leakage of IFNγ from the sections. On the basis of the present findings, we conclude that IFNγ immunostaining of cryostat tissue sections may result in significant staining artifacts. As a consequence, IFNγ immunostaining results on cryostat tissue sections should be interpreted with great caution. The performance of double staining is highly recommended, at least for the distinction between “true” IFNγ positivity and nonspecific staining.
Footnotes
Acknowledgements
We wish to express our gratitude to Prof Dr P.P. Tak and Mr T. Smeets (Academic Medical Center, Dept. of Rheumatology) for providing us with synovial tissue from patients with rheumatoid arthritis.