
Abstract
Our earlier electron microscopic observations revealed that prolonged exposure of glutaraldehyde-fixed rat liver sections to buffer solutions induced focal membrane disruptions of peroxisomes with catalase diffusion as shown cytochemically. Recently, it was suggested that 15-lipoxygenase (15-LOX) might be involved in natural degradation of membrane-bound organelles in reticulocytes by integrating into and permeabilizing the organelle membranes, leading to the release of matrix proteins. We have now investigated the localization of 15-LOX and its role in degradation of peroxisomal membranes in rat liver. Aldehyde-fixed liver slices were incubated in a medium that conserved the 15-LOX activity, consisting of 50 mM HEPES–KOH buffer (pH 7.4), 5 mM mercaptoethanol, 1 mM MgCl2, 15 mM NaN3, and 0.2 M sucrose, in presence or absence of 0.5–0.05 mM propyl gallate or esculetin, two inhibitors of 15-LOX. The exposure of aldehyde-fixed liver sections to this medium induced focal disruptions of peroxisome membranes and catalase diffusion around some but not all peroxisomes. This was significantly reduced by both 15-LOX inhibitors, propyl gallate and esculetin, with the latter being more effective. Double immunofluorescent staining for 15-LOX and catalase revealed that 15-LOX was co-localized with catalase in some but not all peroxisomes in rat hepatocytes. By postembedding immunoelectron microscopy, gold labeling was localized on membranes of some peroxisomes. These observations suggest that 15-LOX is involved in degradation of peroxisomal membranes and might have a physiological role in programmed degradation and turnover of peroxisomes in hepatocytes.
(
T
It is well known that cellular proteins have distinct half-lives and that their amount in the cell is subtly regulated by the proteolytic pathway of the ubiquitin–proteasome system (Rechsteiner 1987; Hershko and Ciechanover 1992). However, the proteins contained in organelles also have their own half-lives, although it is still unclear how the half-life of organelle proteins is regulated. The difference in half-life of organelle proteins could not be explained simply by the autophagic degradation pathway, because in that case all organelle proteins would be degraded simultaneously in autophagolysosomes. On the other hand, if the organelle proteins are degraded within each organelle, the organelle must have its own proteolytic system. Indeed, in yeast mitochondria misfolded proteins have been shown to be degraded ATP-dependently by PIM1 protease (Wagner et al. 1994). However, it is not known whether this protease is also involved in the degradation of regular mitochondrial proteins. In addition, in the intermembrane space of rat liver mitochondria, a metallopeptidase has been found that hydrolyzes oligopeptides but has no action on proteins (Serizawa et al. 1995). Moreover, in peroxisomes, a metalloprotease, an insulin-degrading enzyme, has been identified (Kuo et al. 1994), although its function for degradation of peroxisomal proteins is not known.
In our earlier studies we observed a remarkable heterogeneity in stability of peroxisome membranes in rat liver after exposure of aldehyde-fixed sections to various buffer solutions (Fahimi 1974; Yokota and Fahimi 1978). Thus, the limiting membranes of some peroxisomes were focally disrupted and catalase was confirmed to diffuse out of the peroxisomes by the alkaline DAB staining for this enzyme activity. The biological significance of this observation remained unknown and it was suggested to be related to the well-known high fragility of peroxisomes and the occurrence of particulate vs soluble forms of catalase (Fahimi 1974). Similar observations were reported earlier by others (Legg and Wood 1970; Rigatuso et al. 1970; Wood and Legg 1970), although Novikoff et al. (1972) considered them to be an artifact due to diffusion of oxidized DAB from peroxisomes. Fahimi (1973) ruled out the diffusion of oxidized DAB and showed that it was the enzyme catalase that diffused out of some peroxisomes that exhibited membrane disruptions.
Recently, van Leyen et al. (1998) showed that 15-lipoxygenase (15-LOX) purified from reticulocytes can bind to organelle membranes and induce the leakage of their contents. This ability of 15-LOX to induce diffusion of contents of membrane-bound vesicles was also demonstrated in vitro with liposomes containing a fluorescent dye. The significance of this process for the degradation of cell organelles in differentiating lens fibers (Bassnett and Beebe 1992; Bassnett and Mataic 1997; Dahm et al. 1997) and in maturation of reticulocytes was suggested (Schewe et al. 1975, 1977). If indeed our observation of focal disruption of peroxisomal membranes and catalase leakage occurring during the exposure of aldehyde-fixed liver slices to buffer solution is induced by 15-LOX, inhibitors of this enzyme should prevent this process. We have now studied the effects of 15-LOX inhibitors on the membrane disruption of peroxisomes in normal rat liver. Moreover, using an antibody to a peptide composed of 13 amino acids at the C-terminus of 15-LOX, we investigated the immunocytochemical localization of 15-LOX in normal rat hepatocytes. The results suggest that 15-LOX is involved in degradation of peroxisomal membranes and that it might have a physiological role in programmed degradation and turnover of peroxisomes in hepatocytes.
Materials and Methods
Preparation of Antibodies
Rabbit antibody to 15-LOX peptide was prepared as follows. A peptide consisting of the 13-amino-acid sequence (YLRPSIVENSVAI) at the C-terminus of rabbit 15-LOX was synthesized and 2 mg of it was conjugated with keyhole limpet hemocyanin (Sigma; St Louis, MO) by m-maleimidobenzoyl-N-hydrosuccinimide ester. The conjugate (300 μg of synthetic peptide) was emulsified with complete Freund's adjuvant and 150 μg of peptide was injected SC into the back of each of two Japanese White rabbits (ca. 4 kg bw). The immunization was carried out four times with intervals of 2 weeks. Two weeks after the last injection, about 50 ml of blood was collected from the ear vein of each rabbit. Specificity of the antibody was tested by dot-blot analysis using peptide-conjugated bovine serum albumin, which was prepared as described above. Antibody reacted with 0.05 μg of peptide. The specific antibody to 15-LOX was purified by affinity column chromatography using a peptide-conjugated CH-Sepharose 4B column (Pharmacia Japan; Tokyo, Japan). A guinea pig anti-catalase antibody was prepared as follows. Rat liver catalase was purified as described previously (Yokota and Fahimi 1981). The purified catalase was emulsified with complete Freund's adjuvant. Two hundred μg of catalase in the emulsion was injected four times SC into the back of each guinea pig at intervals of 2 weeks. Blood was collected 2 weeks after the last injection. Specificity of the antibody was checked by immunoblotting analysis.
Purification of 15-LOX from Rabbit Reticulocyte Lysate
Japanese White rabbits weighing 3–4 kg were used. Anemia was induced by injection of 1.25% acetylphenylhydrazine (Sigma–Aldrich Japan; Tokyo, Japan) and a reticulocyte lysate was prepared by the method of Jackson and Hunt (1983). 15-LOX was purified from the lysate according to Rapoport et al. (1979). The final preparation of the enzyme was analyzed by SDS-PAGE.
Immunocytochemistry for 15-LOX
Immunofluorescent Microscopy. Rat liver was fixed by perfusion with 4% paraformaldehyde in 0.2 M HEPES-KOH buffer (pH 7.4) for 10 min. Small tissue blocks were immersed in 2.3 M sucrose solution overnight. Frozen sections 1 μm thick were cut with an Ultracut R microtome (Reichert; Leiden, Germany) equipped with an FC-4D cryosectioning system. Sections were stained doubly for catalase and 15-LOX. We used guinea pig anti-catalase antibody, which was visualized by Alexa 594-conjugated goat anti-guinea pig IgG (Molecular Probes; Eugene, OR) and affinity-purified rabbit anti-15-LOX, which was visualized by Cy2-conjugated goat anti-rabbit IgG (Jackson Immunoresearch Laboratories; West Grove, PA). Sections were observed in a Zeiss Axioplan fluorescent microscope.
Postembedding Immunoelectron Microscopy. Rat liver was fixed by perfusion with a fixative consisting of 0.25% glutaraldehyde in 0.2 M HEPES-KOH buffer (pH 7.4) for 10 min. Small tissue slices (200 μm thick) of fixed liver were dehydrated in graded ethanol and embedded in LR White at −20C. Thin sections were treated with 0.5% bovine serum albumin for 10 min and incubated in affinity-purified rabbit anti-15-LOX antibody or anti-catalase antibody overnight at 4C, followed by a protein A–gold probe (15 nm in diameter).
Quantitative Analysis of the Gold Labeling for 15-LOX in Subcellular Compartments. After the immunogold staining for 15-LOX, 10 electron micrographs were taken at a magnification of × 10,000 and enlarged four times. For analysis of gold labeling density, the areas of peroxisomes, mitochondria, and cytoplasmic matrix were estimated using a digitizer tablet attached to a computer and gold particles located on those areas were counted. The labeling density was expressed as gold particles per μm2 for each compartment. For analysis of relative gold labeling in unit area of sections, the total number of gold particles located over each compartment that was contained in a 100-μm2 area of a section were counted.
Pre-embedding Immunoelectron Microscopy of Light Mitochondrial Fraction. Rat liver was homogenized in a medium containing 300 mM mannitol, 1 mM EGTA, and 10 mM HEPES–KOH (pH 7.2) using a Potter-Elvehjem homogenizer. The homogenate was centrifuged at 5900 × g for 10 min. The supernatant was centrifuged at 25,400 × for 20 min to isolate a light mitochondrial fraction (LMF). The LMF was washed by centrifugation, suspended in the homogenization medium, and mixed with the same volume of 8% paraformaldehyde in 0.4 M HEPES–KOH buffer (pH 7.4). After 20 min the mixture was centrifuged at 10,000 × g for 10 min. A small portion of the resulting pellet was suspended in 0.1 M Tris-HCl buffer (pH 7.4) containing 0.2 M sucrose and centrifuged again. The pellet was suspended in the same buffer containing affinity-purified anti-15-LOX antibody (1 mg/ml) and incubated overnight at 4C, followed by centrifugation and incubation with protein A–gold (15 nm in diameter). After washing with PBS, the LMF pellet was fixed with 2% glutaraldehyde for 20 min, followed by postfixation with 1% reduced osmium. Finally, the pellet was dehydrated and embedded in Epon.
Experiments on Peroxisomal Membrane Disruption
Tissue Fixation. Male Wistar albino rats weighing 180–220 g were used. The liver was fixed by perfusion with a fixative consisting of 4% paraformaldehyde (or 0.25% glutaraldehyde), 15 mM NaN3, in 0.2 M HEPES–KOH buffer (pH 7.4) for 10 min at room temperature (RT), followed by 2-min perfusion with 0.9% NaCl to wash out the excess fixative. The fixed liver was cut into 100-μm slices and further divided into small pieces (1 mm × 1 mm).
Incubation of Liver Tissue Slices. Small tissue slices of fixed liver were incubated in the incubation medium with constant shaking for 4–24 hr at 10C. The incubation medium, which could conserve 15-LOX activity (Rapoport et al. 1979), consisted of 50 mM HEPES–KOH buffer (pH 7.4), 5 mM mercaptoethanol, 1 mM MgCl2, 15 mM NaN3, 0.2 M sucrose, in the presence or absence of 0.5–0.05 mM propyl gallate or esculetin, both of which are 15-LOX inhibitors. After incubation, some tissue slices were washed in 0.9% NaCl and fixed again with 2% glutaraldehyde in 0.1 M HEPES–KOH buffer, pH 7.4, followed by 1-hr postfixation with 1% reduced osmium. After glutaraldehyde fixation, some tissue slices were incubated with alkaline DAB medium for catalase staining, followed by 1% reduced osmium.
Alkaline DAB Reaction for Peroxidase Activity of Catalase. To stain for catalase activity, rat liver was perfusion-fixed with a fixative consisting of 4% paraformaldehyde, 0.25% glutaraldehyde, 0.1 M HEPES–KOH buffer (pH 7.4). Liver slices (100 μm) were incubated for 16 hr at 10C in the same incubation medium indicated above for conservation of 15-LOX activity with or without the specific 15-LOX inhibitors. The liver slices were fixed again in 2% glutaraldehyde for 30 min, followed by 1-hr incubation in the alkaline DAB medium (Fahimi 1969; LeHir et al. 1979).
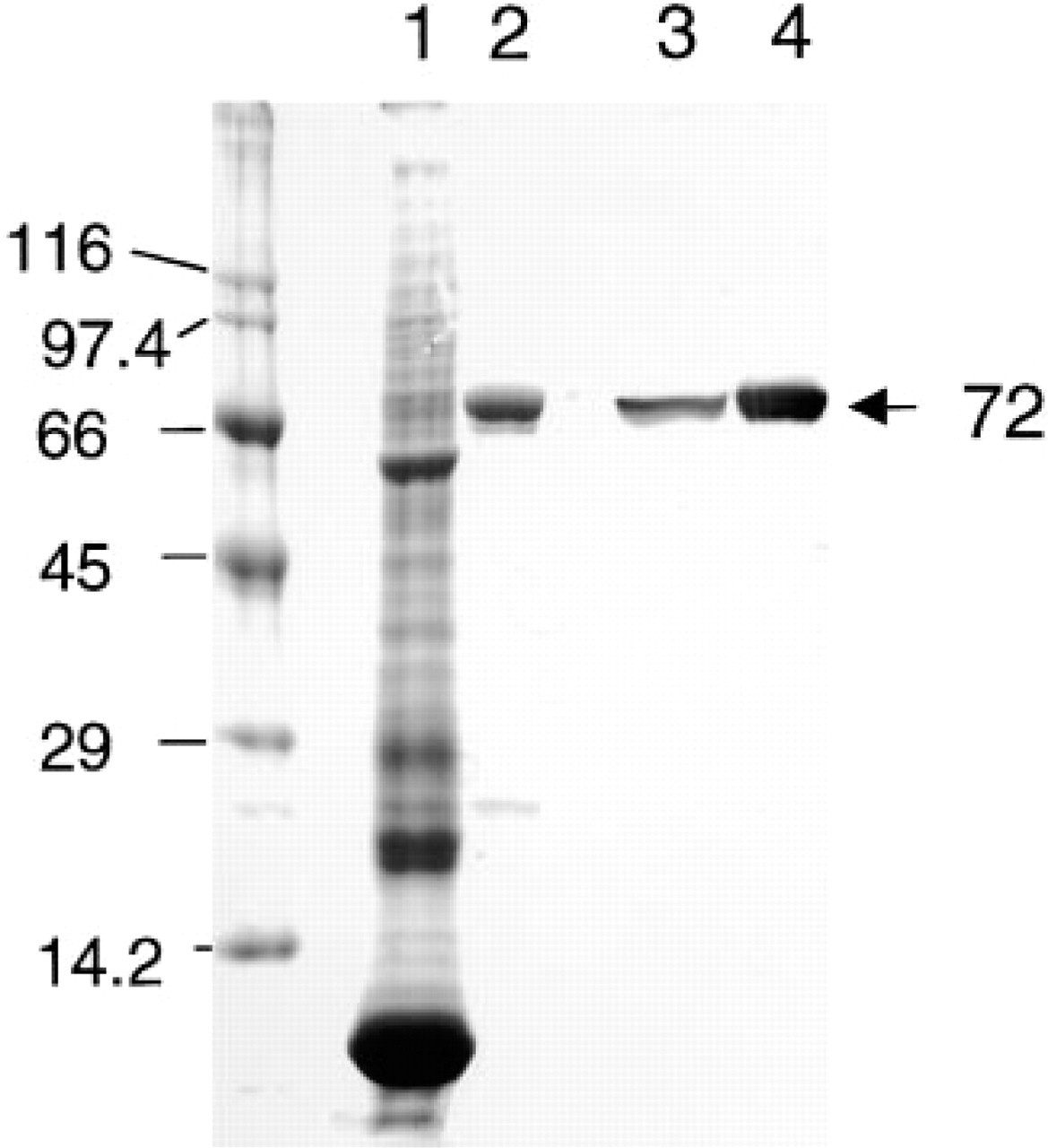
SDS-PAGE and immunoblotting analysis of purified rabbit reticulocyte 15-LOX. Lanes 1 and 2, Coomassie Blue staining; Lanes 3 and 4, immunoblotting. Lanes 1 and 3, rabbit reticulocyte lysate; Lanes 2 and 4, purified rabbit reticulocyte 15-LOX. Numbers at left show the molecular masses of standard proteins and number at right (72 kD) is the molecular mass of 15-LOX.
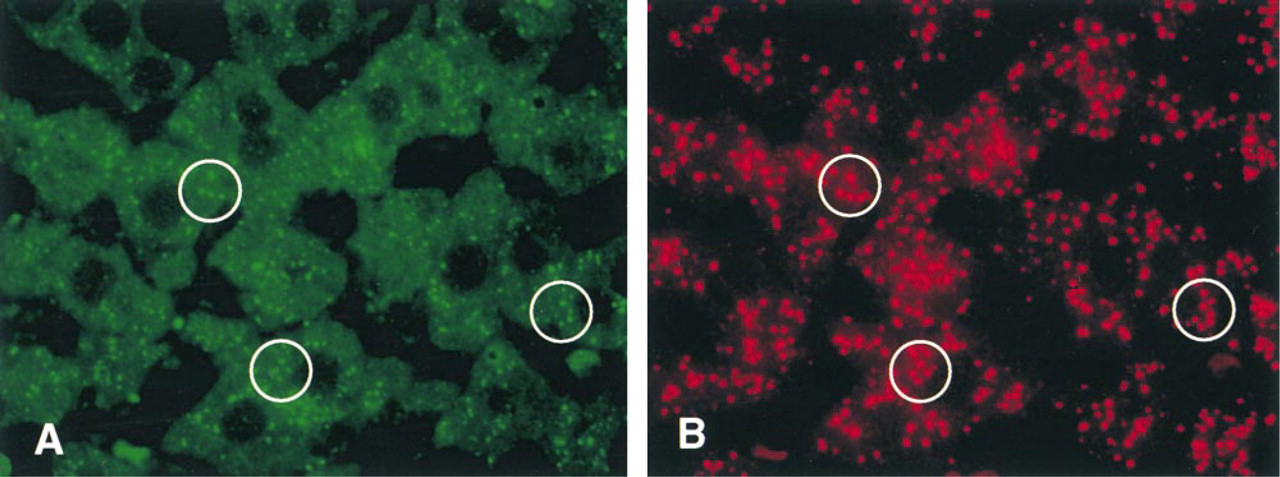
Double immunofluorescent staining of 15-LOX (
Analytical Procedures
Lipoxygenase activity was assayed according to Rapoport et al. (1979). The reaction mixture contained 0.53 mM linoleic acid (Sigma-Aldrich Japan), 0.2% sodium cholate, 5% ethanol in 0.1 M potassium phosphate buffer, pH 7.4. Increase in absorbance at 234 nm (△ A234/min) was monitored with a Beckman DU-20 Spectrophotometer at 2C. Protein was determined by Bradford's method using bovine serum albumin as a standard (Bradford 1976).
Results
Purity of 15-LOX Preparation
The final 15-LOX preparation showed in SDS-PAGE a single band with a molecular mass of 72 kD corresponding to that of 15-LOX (Figure 1), thus confirming the purity of our preparation used for raising the antibody to 15-LOX.
Specificity of the Antibody to 15-LOX Peptide
By immunoblotting, a single band was observed in reticulocyte lysate and purified 15-LOX (Figure 1), showing the monospecificity of the anti-15-LOX antibody.
Double Immunofluorescent Staining for Catalase and 15-LOX
A red color revealing the antigenic sites for catalase was localized to peroxisomes of hepatocytes (Figure 2B). When the same section was treated with rabbit anti-15-LOX antibody followed by Cy2-labeled goat anti-rabbit IgG, a green color showing 15-LOX antigenic sites was observed in some but not all peroxisomes (Figure 2A). In addition, the cytoplasmic matrix also showed moderate green fluorescence. When preimmune serum was used instead of the primary specific antibody, no staining was observed (not shown).
Postembedding Immunoelectron Microscopy of 15-LOX
Gold particles representing 15-LOX antigenic sites were localized in the matrix and membranes of some but not all peroxisomes and mitochondria and in the cytoplasmic matrix (Figure 3). The labeling intensity was relatively low and heterogeneous, with many peroxisomes and mitochondria being negative. Some gold particles were clearly associated with the limiting membranes of peroxisomes and the outer membrane of mitochondria (Figure 3). No gold particles were seen in the sections incubated with preimmune serum followed by protein A–gold probe (not shown). Sections incubated with the antibody to catalase (positive control) showed gold labeling exclusively over the peroxisome matrix (Figure 3 inset), thus confirming the specificity and suitability of our protocol for detection of antigens by immunoelectron microscopy.
Quantitative Analysis of 15-LOX Labeling in Subcellular Compartments
The results are shown in Figure 4. The labeling density (gold particles/μm2) for different subcellular compartments showed significant differences, with peroxisomes exhibiting the highest and the cytoplasm the lowest values and the mitochondria being in between (Figure 4A). On the other hand, when the total number of gold particles in a 100-μm2 area was counted and the distribution in the same three compartments mentioned above was assessed, the cytoplasmic matrix contained 53%, followed by mitochondria (27%) and peroxisomes (20%) (Figure 4B).
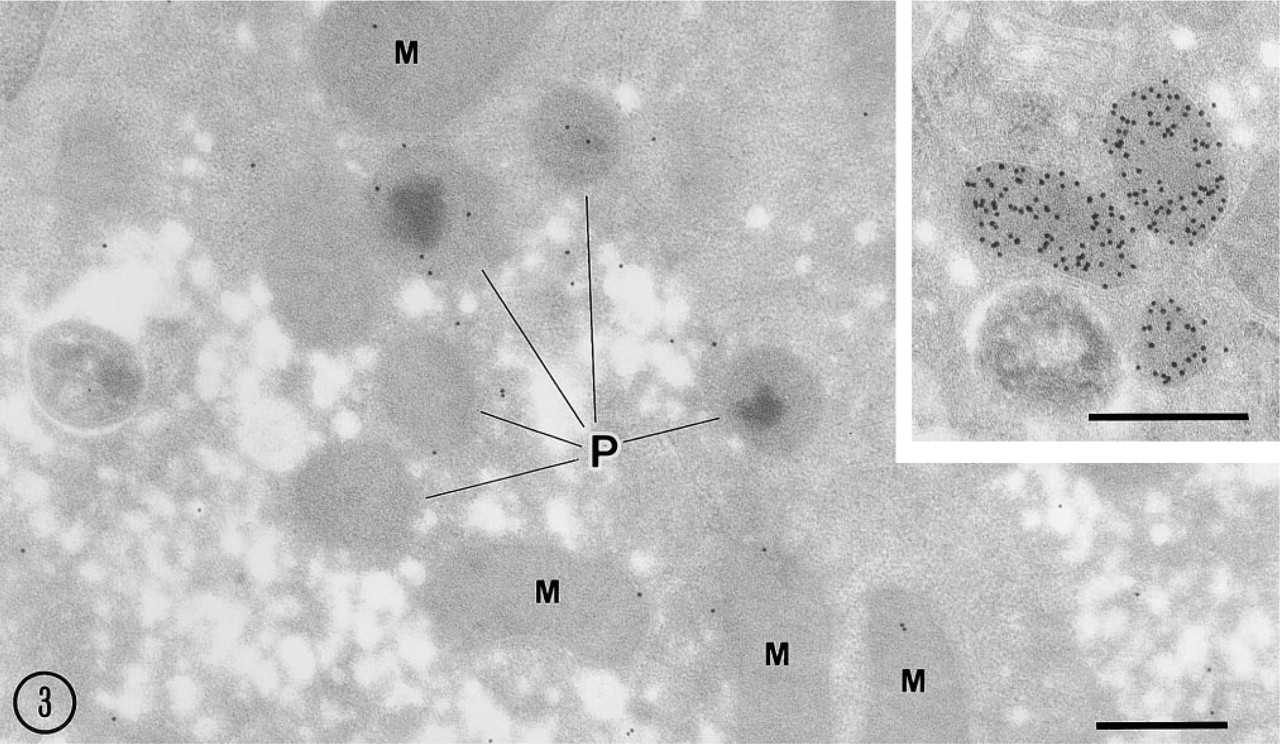
Postembedding electron microscopy of 15-LOX and catalase. Gold particles showing 15-LOX antigenic sites are observed in peroxisomes (P), mitochondria (M), and cytoplasmic matrix. Note that gold particles are associated with membranes of some peroxisomes and mitochondria. (
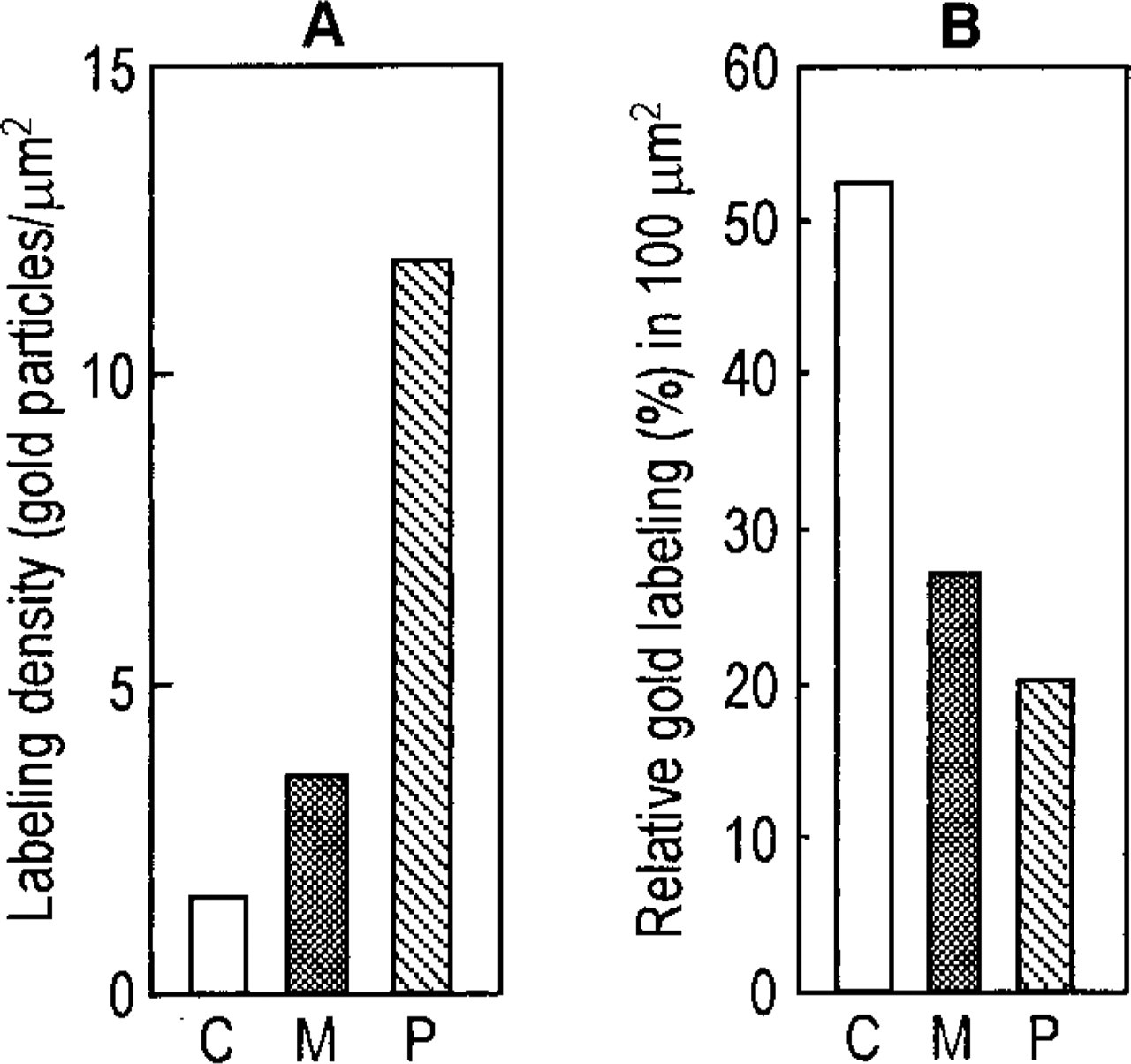
Quantitative analysis of labeling for 15-LOX. (
Pre-embedding Immunoelectron Microscopy of Light Mitochondrial Fraction for 15-LOX
The signal for 15-LOX was associated with the limiting membranes of some peroxisomes and mitochondria as well as with some small vesicles that were free of ribosomes or clathrin coating on their cytoplasmic surface (Figure 5). Essentially, the labeling intensity was low. No gold particles were found in the sample incubated with the preimmune serum followed by the protein A–gold (not shown).
Exposure of Mildly Fixed Liver Slices to Medium Conserving 15-LOX Activity
When 4% paraformaldehyde-fixed liver slices were incubated for 4–24 hrs at 4C in the medium conserving 15-LOX activity, focal disruptions of peroxisomal membrane were observed starting from 8 hr (Figure 6A) but not earlier (not shown). At higher magnification, the peroxisomal membranes were clearly disrupted, exhibiting focal discontinuities (Figure 7A). This process was highly selective, affecting only some but not all peroxisomes. At 12 hr after incubation, the membrane disruption also involved the mitochondrial outer membrane. The process of membrane disruption progressed, and at 24 hr only small pieces of membranes surrounded some peroxisomes. Membranes of the ER and mitochondria were also heavily disrupted at 24 hr after incubation (not shown).
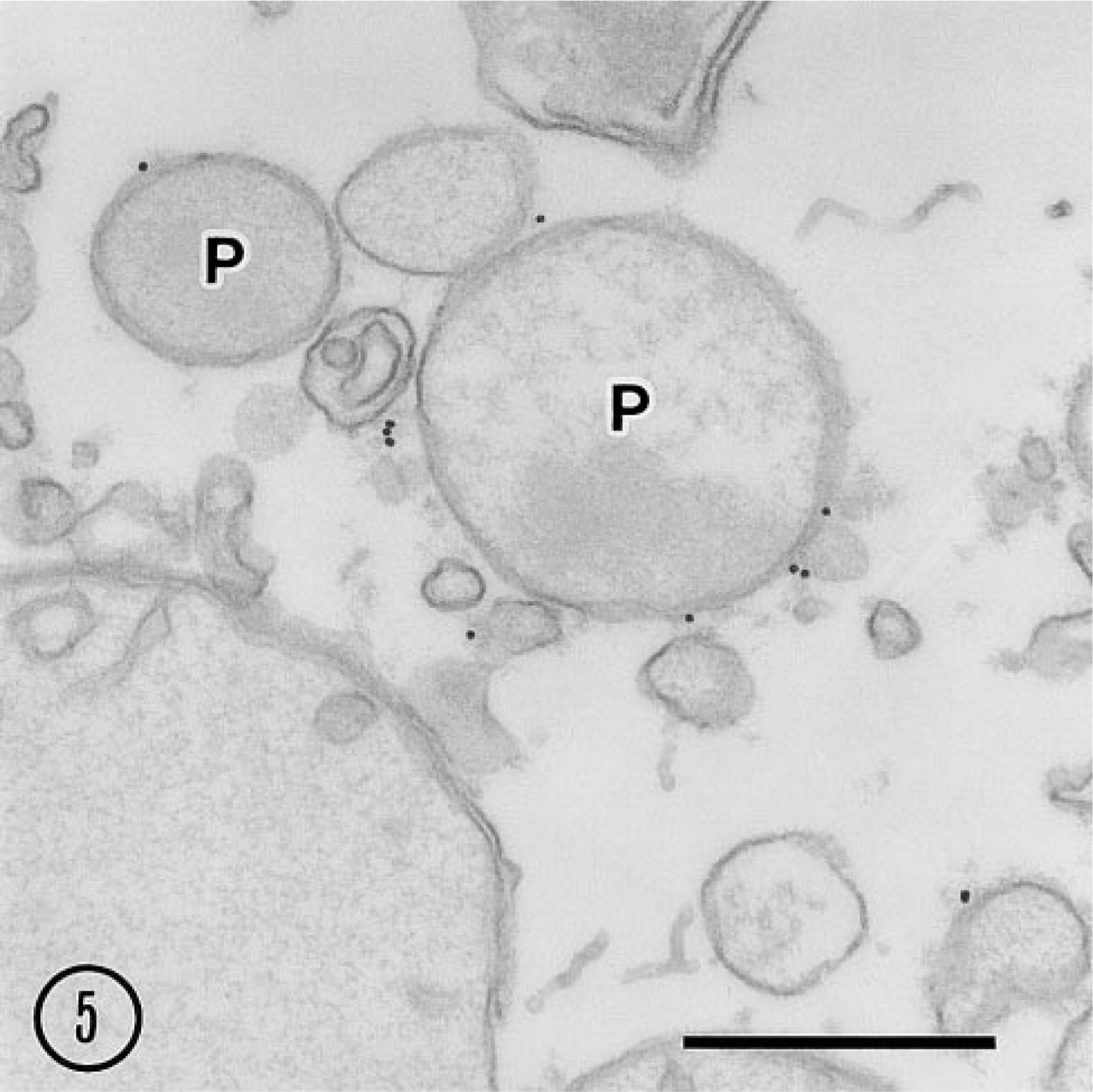
Pre-embedding immunoelectron microscopy of light mitochondrial fraction. Gold particles showing 15-LOX antigenic sites are associated with peroxisome (P) membrane and other membrane-bound vesicles. Bar = 0.5 μm.
Prevention of Membrane Disruption by 15-LOX Inhibitors
When inhibitors for 15-LOX, propyl gallate and esculetin, were present in the incubation medium, the membrane disruption mentioned above was significantly reduced (Figures 6 and 7). Both inhibitors showed their effects at a concentration of 50 μM. The protective effect of esculetin was stronger than that of propyl gallate. Propyl gallate could not inhibit the membrane disruption of some peroxisomes in all time courses, and particularly after 24 hr of incubation the disruption was noted in almost all peroxisomes (not shown). Esculetin could for the most part reduce the membrane disruption until 12 hr, but some peroxisomes showed focal breakdown of their membranes after 24-hr incubation (not shown).
Diffusion of Catalase from Peroxisomes with Focal Membrane Disruption
To investigate the relationship between the diffusion of catalase and membrane disruption, we used glutaraldehyde fixation instead of paraformaldehyde alone, as used for our immunocytochemical studies. Glutaraldehyde fixation delayed membrane disruption of peroxisomes compared to paraformaldehyde. When slices of rat liver fixed with 0.25% glutaraldehyde were exposed for 16 hr to the incubation medium preserving the activity of 15-LOX, clear evidence of diffusion of catalase, as revealed by the DAB reaction, was detected around some peroxisomes (Figure 8A). This diffusion of catalase was mostly prevented when esculetin, an inhibitor of 15-LOX, was present in the incubation medium (Figure 8B). Tissue slices incubated for 12 hr showed minimal or no diffusion of catalase.
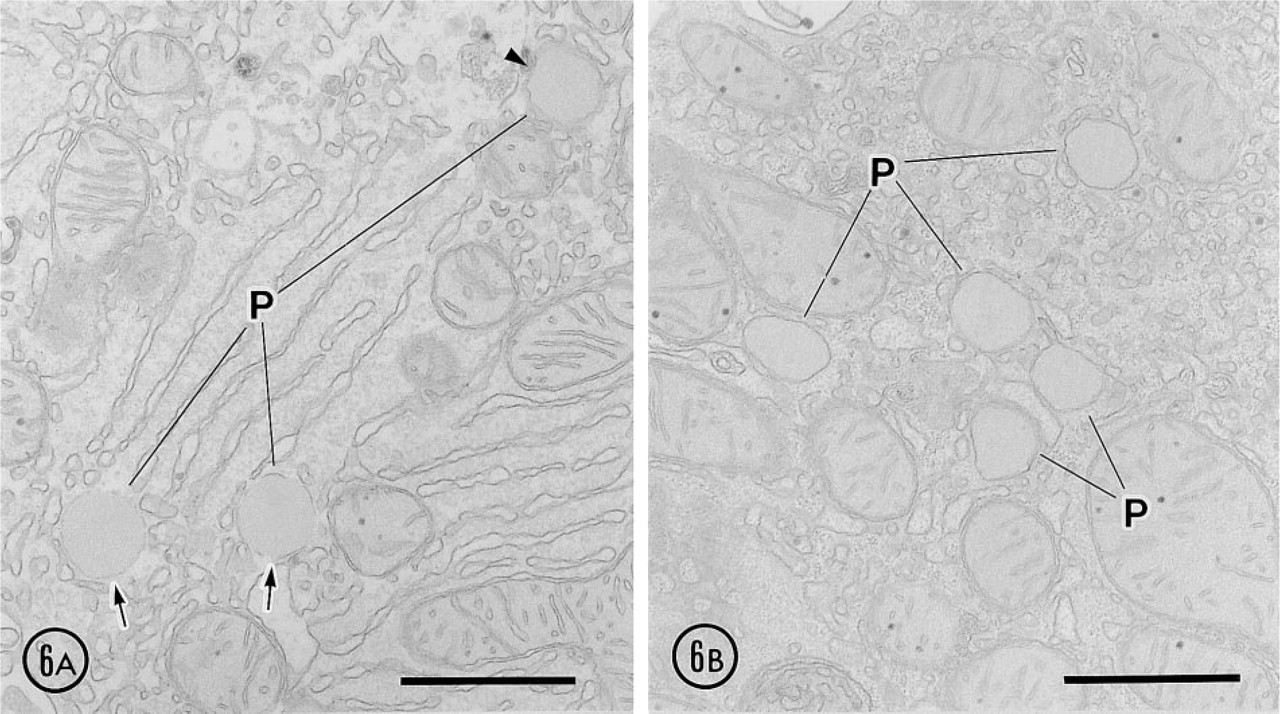
Effect of 15-LOX inhibitors after 8-hr incubation. (
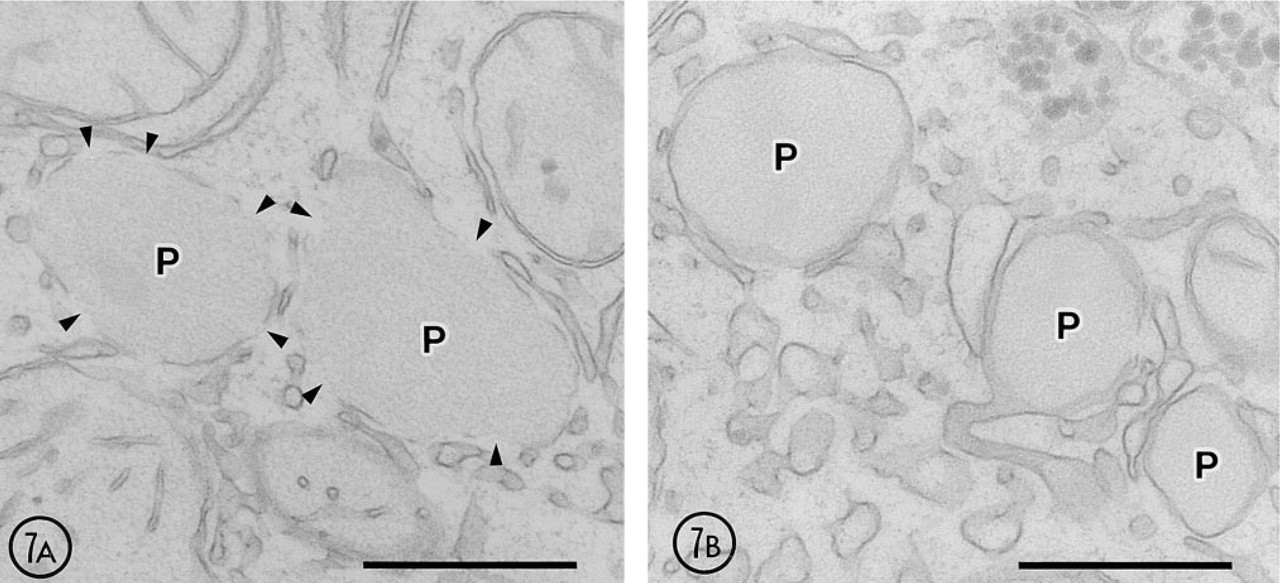
High-power view showing the effect of 15-LOX inhibitors. (
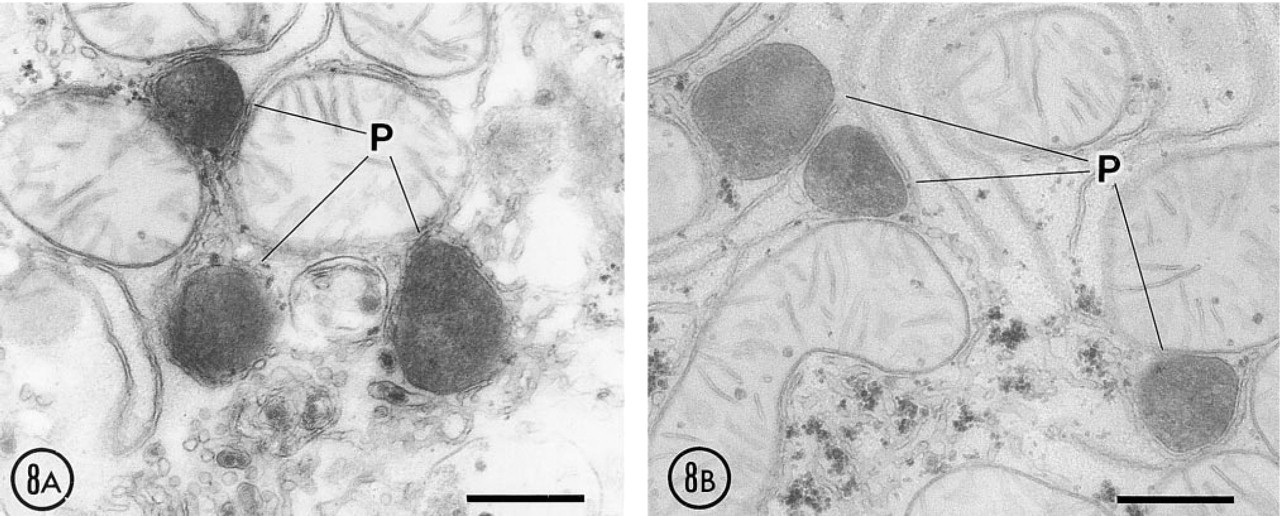
Effect of 15-LOX inhibitors after 16-hr incubation. Liver was fixed with 0.25% glutaraldehyde. (
Discussion
Immunocytochemical Localization of 15-LOX
In the present study, double immunofluorescent staining of normal rat liver for calatase and 15-LOX showed that 15-LOX was localized in peroxisomes and in the cytoplasmic matrix and that this enzyme was associated with some but not all peroxisomes (Figure 2). The mitochondrial staining for 15-LOX by immunofluorescence was significantly weaker than that of peroxisomes and was submerged in the diffuse cytoplasmic staining so that individual mitochondria were not visualized. The prominent peroxisomal staining was also confirmed by postembedding immunoelectron microscopy (Figure 3), which revealed gold labeling for 15-LOX in association with the membranes of a limited number of peroxisomes. Furthermore, pre-embedding immunoelectron microscopy confirmed that gold labeling for 15-LOX was associated with the peroxisomal membranes (Figure 5). These results clearly show that 15-LOX binds to the membranes of some but not all peroxisomes in hepatocytes of normal rat. This supports the recent biochemical findings of van Leyen et al. (1998), who showed that 15-LOX purified from rabbit reticulocytes could bind to the membranes of some organelles such as mitochondria and ER. Indeed, our immunoelectron microscopic observations revealed that signals for 15-LOX were associated with some of those organelles. Preembedding immunoelectron microscopy of the LMF showed that membranes of some “smooth vesicles” were also labeled for 15-LOX. Similar results were reported by Brinckmann et al. (1998). The present results strongly suggest that 15-LOX is associated with the cytoplasmic surface of membranes of organelles such as peroxisomes, mitochondria, and other membrane-bound compartments. Quantitative analysis of immunolabeling for 15-LOX showed that this enzyme distributed in peroxisomes at the highest concentration. However, when the ratio of gold labeling in subcellular compartments to the total labeling in 100 μm2 was calculated, approximately 53% of gold particles representing 15-LOX was found in the cytoplasm, indicating that there is a large cytoplasmic pool of 15-LOX, in addition to its membrane-associated form in hepatocytes.
Membrane Disruption of Peroxisomes of Mildly Fixed Liver Tissue
Our early observations showed that when chopper or microslicer sections of rat liver fixed with 2.5% glutaraldehyde were exposed to a buffer solution for 2 days at 4C, focal membrane disruption with catalase diffusion occurred in some but not all peroxisomes (Fahimi 1973, 1974). Assuming that this phenomenon could be due to an enzymatic process, we used in the present study a milder fixation, with 4% paraformaldehyde or 0.25% glutaraldehyde. Furthermore, to assess the possible involvement of 15-LOX in the membrane disruption process, we used a medium that could conserve the activity of this enzyme (Rapoport et al. 1979). By using this combination, we could shorten the time required for the focal disruptions of the peroxisomal membrane to only 8 hr. Moreover, the present study showed that fixation with 0.25% glutaraldehyde delayed the start of membrane disruption compared with 4% formaldehyde. Although the temperature is an important factor in enzymatic reactions and higher temperatures accelerate the reaction velocity, the activity of purified 15-LOX is lost quickly at normal RT, and indeed the activity of that enzyme is usually measured at 2C (Rapoport et al. 1979). We therefore chose 10C for the incubation of sections to assess the role of 15-LOX in the present study.
Inhibitors of 15-LOX Prevent Membrane Disruption
A major finding of the present study is that inhibitors of 15-LOX, propyl gallate and esculetin, significantly reduced the membrane disruptions of peroxisomes during exposure of aldehyde-fixed rat liver sections to the incubation medium. The inhibitors we used are known to prevent the binding of purified 15-LOX to the membranes of isolated organelles (van Leyen et al. 1998). This was also confirmed by the experiments in which the diffusion of catalase from peroxisomes, as detected by the alkaline DAB reaction, was mostly prevented in the presence of esculetin. Taken together, our results strongly suggest that peroxisomal membrane disruption during the incubation is caused by the activity of 15-LOX (or a similar enzyme) which is inhibited by 15-LOX inhibitors. The membrane disruption was inhibited only partially by propyl gallate and more effectively by esculetin. This differential effect further supports the notion that the focal disruption of peroxisomal membranes was due to the function of the enzyme 15-LOX. Furthermore, the membrane disruption occurred in some but not all peroxisomes. This selective occurrence of membrane disruption implies that some peroxisomes are selected to be degraded, although the exact mechanism of this process is not known.
In the steady-state situation, the number of organelles, including peroxisomes, in a cell is maintained at a constant level (Sakai et al. 1998). The peroxisomes induced by various peroxisome proliferators are rapidly destroyed when the proliferating stimuli are discontinued (Moody and Reddy 1976). The present results suggest that 15-LOX might be involved in such a degradation process and in regulation of the organelle number in hepatocytes. In our experiments, however, we used aldehyde-fixed tissue, which is obviously not comparable with the in vivo situation. Therefore, it is important to investigate in the future the effects of 15-LOX on peroxisomes and other organelles in living cells or in semipermeabilized cells.
In conclusion, our observations suggest that 15-LOX is involved in degradation of peroxisomal membranes and that it could have a physiological role in programmed degradation and turnover of peroxisomes in hepatocytes.
Footnotes
Acknowledgements
Acknowledgments
Supported in part by grants-in-aid for scientific research no. 03833013 to Sadaki Yokota, by the Naito Foundation, Japan, and by the Shizuoka Research Institute, Japan, to Toshiaki Oda. The work in Heidelberg University is supported by SFB 601, Project B 1, and FA-146 of the Deutsche Forschungsgemeinschaft to H.D. Fahimi.
We thank Annemarie Achten for secretarial help and word processing.