
Abstract
Transamination of branched-chain amino acids (BCAAs) catalyzed by the branched chain aminotransferase isoenzymes (BCATs) is believed to play an important role in nitrogen shuttling and excitatory neurotransmitter glutamate metabolism in brain. Recently, we have shown that the mitochondrial isoenzyme (BCATm) is the predominant form found in cultured astrocytes. In this study we used immunocytochemistry to examine the distribution of BCAT isoenzymes in cultured rat neurons and microglial cells. The cytoplasm of neurons displayed intense staining for the cytosolic isoenzyme (BCATc), whereas BCATm staining was not detectable in neurons. In contrast, microglial cells expressed BCATm in high concentration. BCATc appeared to be absent in this cell type. The second and committed step in the BCAA catabolic pathway is oxidative decarboxylation of the α-keto acid products of BCAT catalyzed by the branched-chain α-keto acid dehydrogenase (BCKD) enzyme complex. Because the presence of BCKD should provide an index of the ability of a cell to oxidize BCAA, we have also immunocytochemically localized BCKD in neuron and glial cell cultures from rat brain. Our results suggest ubiquitous expression of this BCKD enzyme complex in cultured brain cells. BCKD immunoreactivity was detected in neurons and in astroglial and microglial cells. Therefore, the expression of BCAT isoenzymes shows cell-specific localization, which is consistent with the operation of an intercellular nitrogen shuttle between neurons and astroglia. On the other hand, the ubiquitous expression of BCKD suggests that BCAA oxidation can probably take place in all types of brain cells and is most likely regulated by the activity state of BCKD rather than by its cell-specific localization.
Keywords
T
The second reaction in BCAA degradation is the oxidative decarboxylation of the branched chain α-keto acids (BCKAs) catalyzed by the mitochondrial branched chain α-keto acid dehydrogenase (BCKD) enzyme complex. Because this step is essentially irreversible, passage of the α-keto acids corresponding to the BCAAs through this step commits the carbon skeleton to degradation. The multienzyme complex consists of three different subunits, E1, E2, and E3, and is structurally related to the pyruvate dehydrogenase enzyme complex (Pettit et al. 1978; Danner et al. 1979). The enzymatic activity of BCKD is regulated by phosphorylation–dephosphorylation (Lau et al. 1982; Harris et al. 1986). Knowledge of the cellular distribution of BCKD among neural cells would allow identification of the cell types in brain that have the capacity to oxidize the BCAAs. These results may provide understanding of brain abnormalities found in disorders of BCAA metabolism such as maple syrup urine disease.
BCAAs are believed to be involved in brain glutamate metabolism, especially because Kanamori et al. (1998) have reported that leucine can provide about 25% of brain glutamate nitrogen. Yudkoff (1997) and others (Bixel et al. 1997) have proposed that BCAAs and BCKAs are involved in a nitrogen shuttle between astrocytes and neurons. Recently, it has been hypothesized that BCAAs provide the amino group for optimal rates of de novo glutamate synthesis via BCATm in astrocytes (Hutson et al. 1998). The uptake of BCAA then promotes glutamine transfer to neurons, where BCATc reaminates the BCKA in neurons for another cycle (Bixel et al. 1997; Hutson et al. 1998). In agreement with our previous work (Bixel et al. 1997), immunoblots of cultured brain cell extracts using BCATm- and BCATc-specific antibodies revealed that, over time, BCATm becomes the predominant BCAT in astroglial cultures, whereas BCATc is the predominant isoenzyme found in early neuron cultures (Hutson et al. 1998). These results suggested that this isoenzyme may be located in neurons. However, these cultures also contained contaminating glial cells. Therefore, in this study we have used immunocytochemistry to identify the cellular distribution of BCAT isoenzymes in neuron-rich primary cultures and in glial cell cultures from rat brain. To determine the site of BCKA oxidation, we have also examined the cellular localization of BCKD in the primary cultures. The results suggest that in the primary cultures BCATc is expressed in neurons, whereas microglial cells express BCATm. BCKD is found in both neurons and glial cells.
Materials and Methods
Materials
Dulbecco's modified Eagle's medium (DMEM) and fetal calf serum (FCS) were obtained from GIBCO (Eggenstein, Germany). All cell culture plasticware was from Nunc (Wiesbaden, Germany). Acrylamide and N,N′-methylene bisacrylamide were obtained from Bio-Rad Laboratories (München, Germany). 5-Bromo-4-chloro-3-indolyl-phosphate and 4-nitroblue tetrazolium chloride, premixed protein molecular weight markers (cat. no. 1495984), sodium dodecyl sulfate (SDS), and Tris were from Boehringer (Mannheim, Germany). N,N-N′,N′-tetramethyl-ethylendiamine, Triton X-100, and Tween-20 were from Serva (Heidelberg, Germany). Nitrocellulose filter sheets were purchased from Millipore (Eschborn, Germany). Bovine serum albumin (BSA), poly-
Cell Culture
Neuron-rich primary cultures were prepared from the brains of 16-day-old rat embryos as described (Löffler et al. 1986; Pfeiffer et al. 1989). Neuron-rich primary cultures were used for immunostaining on Day 5. Astroglia-rich primary cultures were derived from the brains of newborn Wistar rats, cultured as described previously (Hamprecht and Löffler 1985), and used for immunostaining on Day 7. Microglial secondary cultures were obtained from astroglia-rich primary cultures cultivated for 14 days in culture flasks. In culture, the vast majority of microglial cells sit on top of a confluent cell layer of astroglial cells and can be removed by shaking the culture flask (Graeber et al. 1989). The culture medium with the floating microglial cells was removed and the cells were concentrated by centrifugation. Microglial cells were resuspended in culture medium and seeded in culture dishes. Non-adherent cells were discarded by renewing the culture medium after 1 hr. The purity of the microglial secondary cultures, as assessed by counting the number of OX-42 positive cells, was routinely 98–99%. After 3-hr incubation at 37C in a humidified atmosphere of 10% CO2/90% air, the cells were used for immunocytochemistry. For immunocytochemistry, cells were grown on glass coverslips (22 mm2).
Immunocytochemistry
For BCAT/vimentin, BCAT/OX42, BCKD/GFAP, BCKD/vimentin, and BCKD/OX42 double staining, the cells were fixed at room temperature (RT) using 4% (w/v) paraformaldehyde in PBS, pH 7.4, for 10 min. The cells were washed twice in PBS for 5 min, then once with 0.1% (w/v) glycine in PBS, pH 7.4, for 5 min. Next, the cells were permeabilized with 0.3% (w/v) Triton X-100 in PBS for 10 min. The anti-sera against BCAT isoenzymes and the antivimentin monoclonal antibodies were diluted in PBS containing 0.3% (w/v) Triton X-100 and 10% (v/v) normal goat serum (Protocol A). This staining method proved to be advantageous for detection of the BCAT isoenzymes and BCKD. Because the anti-GAP-43 antibodies were unable to recognize their antigen when the cells were treated according to this procedure, for BCAT/GAP-43 and BCKD/GAP-43 double staining the cells were fixed with 4% (w/v) paraformaldehyde in PBS at pH 7.4 for 15 min. The cells were permeabilized by ice-cold methanol for 5 min using a modification of the method described by Deloulme et al. (1990). The antisera against the BCAT isoenzymes and the anti-GAP-43 monoclonal antibodies were diluted in PBS containing 3% (w/v) BSA (Protocol B). After Protocol B, the labeled cells exhibited somewhat higher background staining (see Figure 2D) than the cells treated according to Protocol A.
For immunostaining of cells on the coverslips, they were placed in a humidified chamber and then exposed to a mixture of the two primary antibodies for 2 hr and separately to the mixture of the two secondary antibodies for 1 hr. The figure legends indicate the corresponding antibody solutions used for the individual double-labeling experiments. The staining of the cultured cells was the same whether they were stained with individual antibodies or with both antibodies in the same cocktail. After each incubation, the cells were washed twice either with 0.1% (w/v) Triton X-100 in PBS for BCAT/vimentin staining or with 0.3% (w/v) BSA in PBS for BCAT/GAP-43 staining. The coverslips were mounted, cells down, using 50% (v/v) glycerol in PBS. The preparations were viewed by glycerol immersion optics using a Zeiss fluorescence microscope (IM35) with a Plan-Neofluar ×25 objective. The specificity of the BCATc, BCATm, and BCKD antisera was evaluated by preabsorbing the antisera with their corresponding antigens before using them for immunocytochemistry. The suitable antigen concentration for preincubation of the antiserum was determined using ELISAs. ELISAs were performed by coating 96-well plates overnight at 4C with 100 ng BCAT. BCATm was purified from rat heart mitochondria (Hall et al. 1993). An E. coli cell extract containing recombinant human BCATc was used as a source of BCATc (Davoodi et al. 1998). Purified rat liver BCKD served as the source of BCKD antigen (Shimomura et al. 1990).
Before use in immunocytochemistry, the BCATm (BCATc) antiserum (diluted 1:200) was preincubated with 8 μg BCATm (5 μg BCATc) in a total volume of 80 μl at 4C for 12 hr. We have shown previously that BCATm is abundant in astroglial cultures (Bixel et al. 1997). Immunocytochemical staining of an astroglia-rich primary culture with BCATm antiserum preabsorbed with BCATm antigen revealed only background staining (Figures 1A and 1B). Because astroglia-rich primary cultures express BCATc, these same cultures were used to test for the specificity of the BCATc antiserum. Background staining of astroglia-rich primary cultures using BCATc antiserum with BCATc antigen preincubation is shown in Figures 1C and 1D. Background staining for BCKD was determined using preabsorbed BCKD antiserum (diluted 1:100). For preabsorption, BCKD antiserum (diluted 1:40) was preincubated with 200 μg BCKD in a final volume of 100 μl. When the preabsorbed BCKD antiserum for staining of astroglia-rich primary cultures was used only background staining of the cells was observed (Figures 1E and 1F).
Immunoblotting
The cell culture homogenates were prepared as described previously (Bixel et al. 1997). SDS-PAGE was performed according to the procedure of Laemmli (1970) using 10% (w/v) acrylamide gels. For Western blotting (Burnette 1981), proteins were transferred onto nitrocellulose at a current of 0.3 A for 12 hr in transfer buffer (25 mM Tris, 192 mM glycine, adjusted to pH 9.0 with KOH). Proteins transferred to nitrocellulose membranes were stained briefly with Ponceau and subsequently de-stained with water The filter was blocked using 1% (w/v) milk powder in incubation buffer [20 mM Tris-HCl, 150 mM NaCl, 0.02% (w/v) Tween-20, pH 7.4] at RT for 2 hr. The filters were washed three times with incubation buffer, then incubated with the BCATm or BCATc IgG fraction (diluted 1:200 in incubation buffer) at RT for 2 hr. Unbound antibodies were removed by washing three times with incubation buffer and the filters were incubated with secondary alkaline phosphatase-conjugated anti-rabbit IgG diluted 1:3000 in incubation buffer. After washing the strips with incubation buffer, color was developed in staining buffer (100 mM Tris-HCl, 50 mM NaCl, 5 mM MgCl2, 365 μM 5-bromo-4-chloro-3-indolyl-phosphate, 45 μM 4-nitroblue tetrazolium chloride, pH 8.9).
Results
To determine the distribution of BCAT isoenzymes in neurons, we have immunocytochemically examined neuron-rich primary cultures prepared from embryonic rat brains using antisera against rat BCATc and rat BCATm. Staining of the cultures for BCATc revealed strong immunoreactivity in cells with small and round somata bearing several thin and extended processes (Figures 2A and 3A). These cells were identified as neurons by double staining with antibodies against the neuronal marker GAP-43 (Figure 2B; Meiri et al. 1988). The neurites reacted strongly with GAP-43 antibodies, whereas the cell bodies were less intensely labeled. The BCATc immunoreactivity in neurons was evenly distributed throughout the entire cell, including the processes (Figures 2A and 3A). All neurons in the culture appeared to be BCATc-positive. Immunocytochemical analysis using BCATm antiserum resulted in only background staining of GAP-43-positive neurons (Figures 2D and 3D). Staining of neuron-rich primary cultures using an anti-vimentin monoclonal antibody, which specifically labels astroglial cells in all developmental stages, was used to identify the astroglial cells in these cultures (Figures 3B and 3E; Dahl et al. 1981). As we have shown previously (Bixel et al. 1997), astroglial cells stained positively for BCATm (Figure 3D). It has been documented that the percentage of astroglial cells present in neuron-rich cultures increases strongly with the age of the culture (Löffler et al. 1986). Nevertheless, no striking differences in intensity of the BCATc immunoreactivity in the neuronal cells were observed with cells cultured for periods of up to 9 days (data not shown), suggesting that expression of the BCATc in neurons is not dependent on the number of astroglial cells present in the culture.
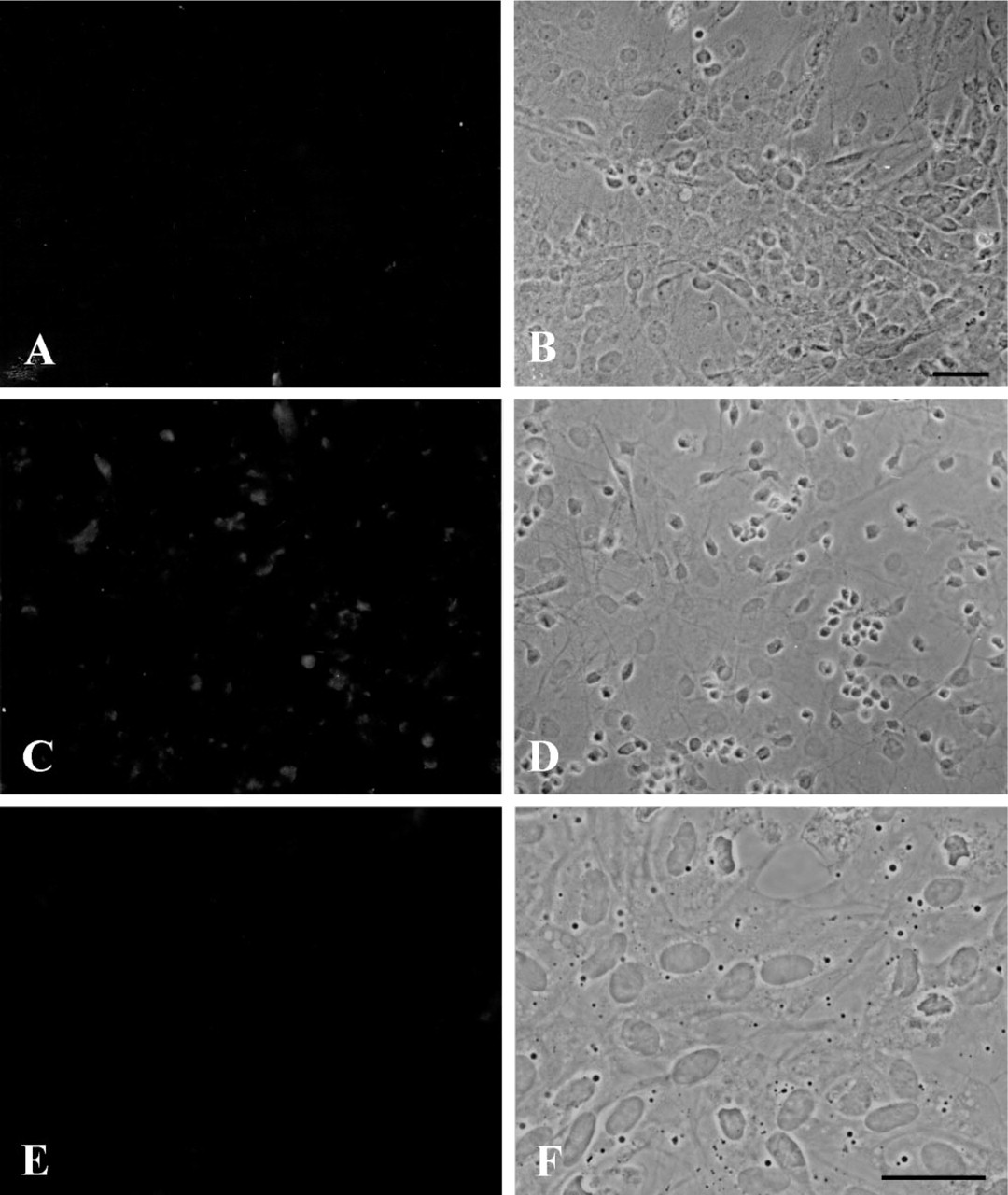
Specificity of the BCATm, BCATc, and BCKD antisera. Astroglia-rich primary cultures were immunocytochemically stained using BCATm (
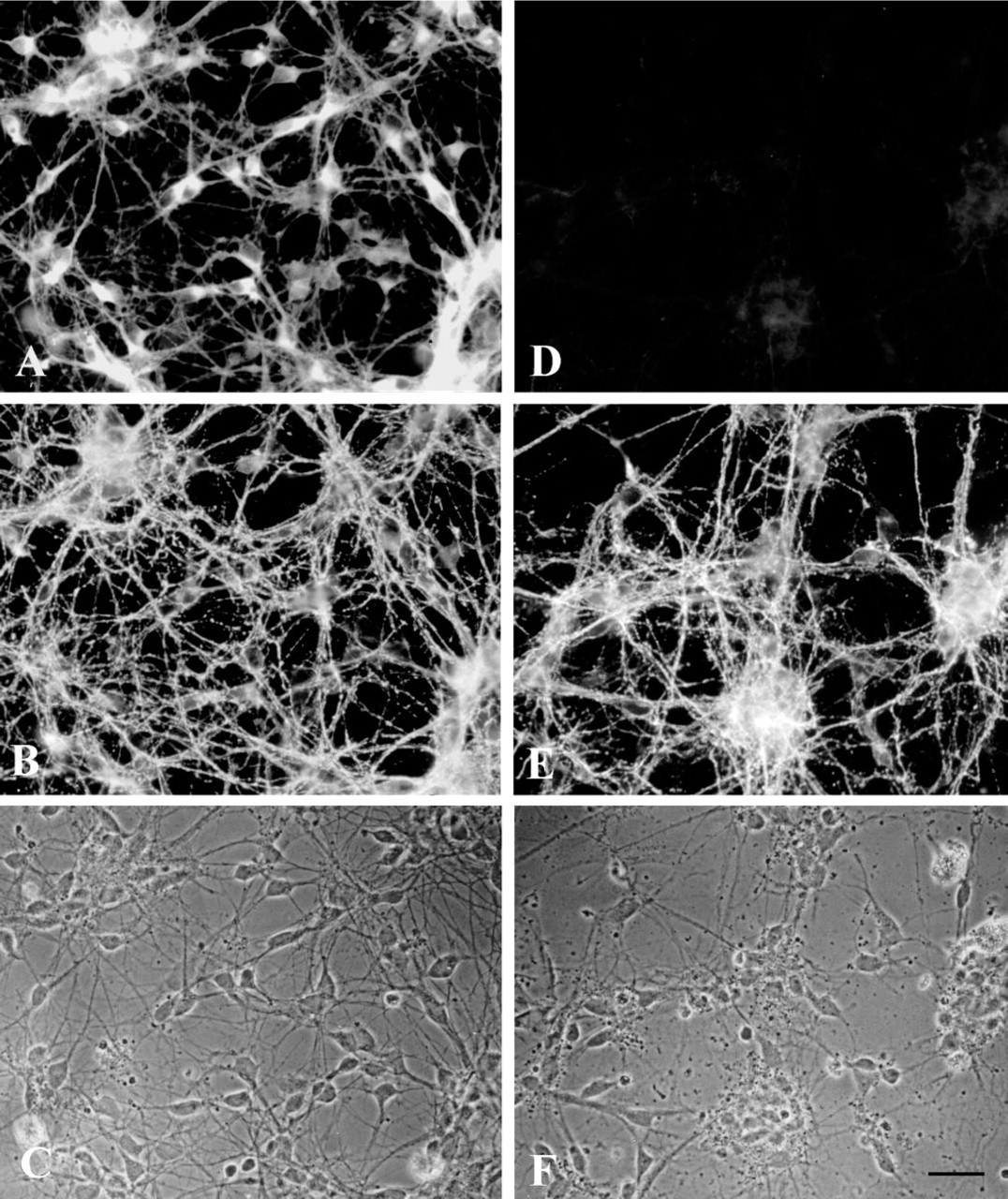
Immunofluorescence staining of neuron-rich primary cultures for the BCAT isoenzymes BCATc (
Immunofluorescence staining of neuron-rich primary cultures for the BCAT isoenzymes BCATc (
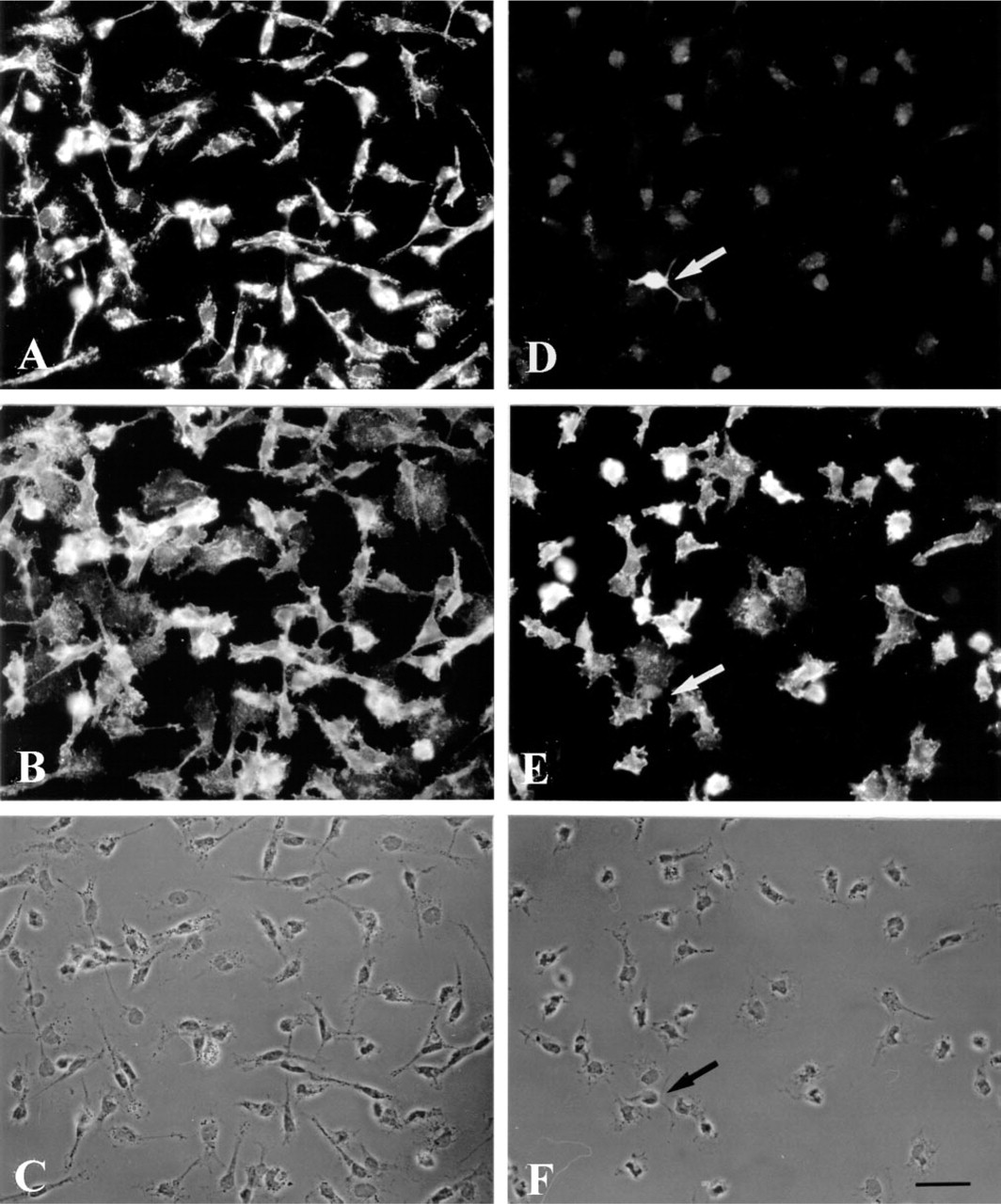
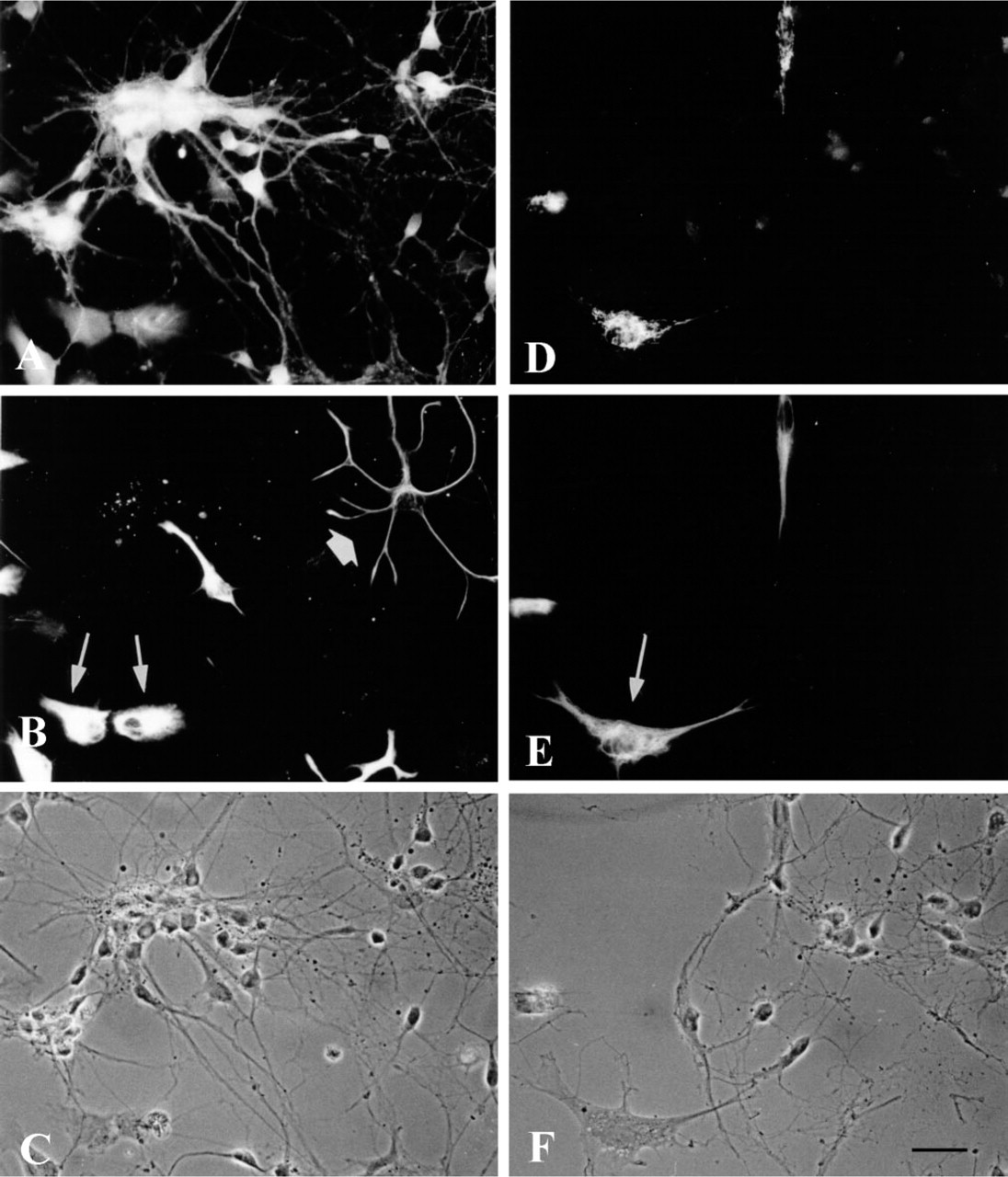
Immunofluorescence staining of microglial secondary cultures for BCATm (
The distribution of both BCAT isoenzymes was also investigated in microglial cells. Astroglia-rich primary cultures, in which about 10% of the cell population are microglial cells, were used as the source to obtain almost pure microglial secondary cultures (Graeber et al. 1989). By shaking the culture flasks, mainly microglial cells were released into the culture medium. Shortly after reseeding, these cells adhered to the culture dish, showing round cell morphology. Then the microglial cells started to develop processes and became irregular in shape. After 3 hr, the main population consisted of flat cells with spinous processes or cytoplasmic extensions with ruffled edges, while other cells showed a more spindle-like appearance (Figure 4). The microglial cells were identified immunocytochemically by the presence of the marker antigen CR3 (complement receptor Type 3), which specifically reveals the presence of both resting and phagocytic microglial cells (Graeber et al. 1989). Practically all cells in the field stained positively for CR3 (Figures 4B and 4E). In general, flat and extended microglial cells were stained less intensely than cells showing a small and round morphology and bearing occasional short processes. Immunofluorescence staining using antiserum against BCATm in OX42-positive microglial cells is shown in Figure 4A. The BCATm staining appeared to be concentrated in the area surrounding the cell nuclei, whereas the nuclei were unstained. In contrast, staining of microglial secondary cultures using BCATc antiserum yielded primarily background immunoreactivity (Figure 4D). As an example, the arrow in Figure 4D points to a cell that stained positively for BCATc. This CR3-negative “contaminating” cell is most likely an oligodendroglial cell (Figure 4E). The presence of BCATm, but not of BCATc, in cultured microglial cells was also confirmed by Western blotting analysis (Figure 5). BCATm antiserum recognized a single protein band corresponding to the molecular mass of 41 kD in microglial cell homogenates (Figure 5, Lane 3). Western blotting analysis using BCATc antiserum did not reveal a 47-kD protein band corresponding to the molecular mass of rat BCATc (Figure 5, Lane 4).
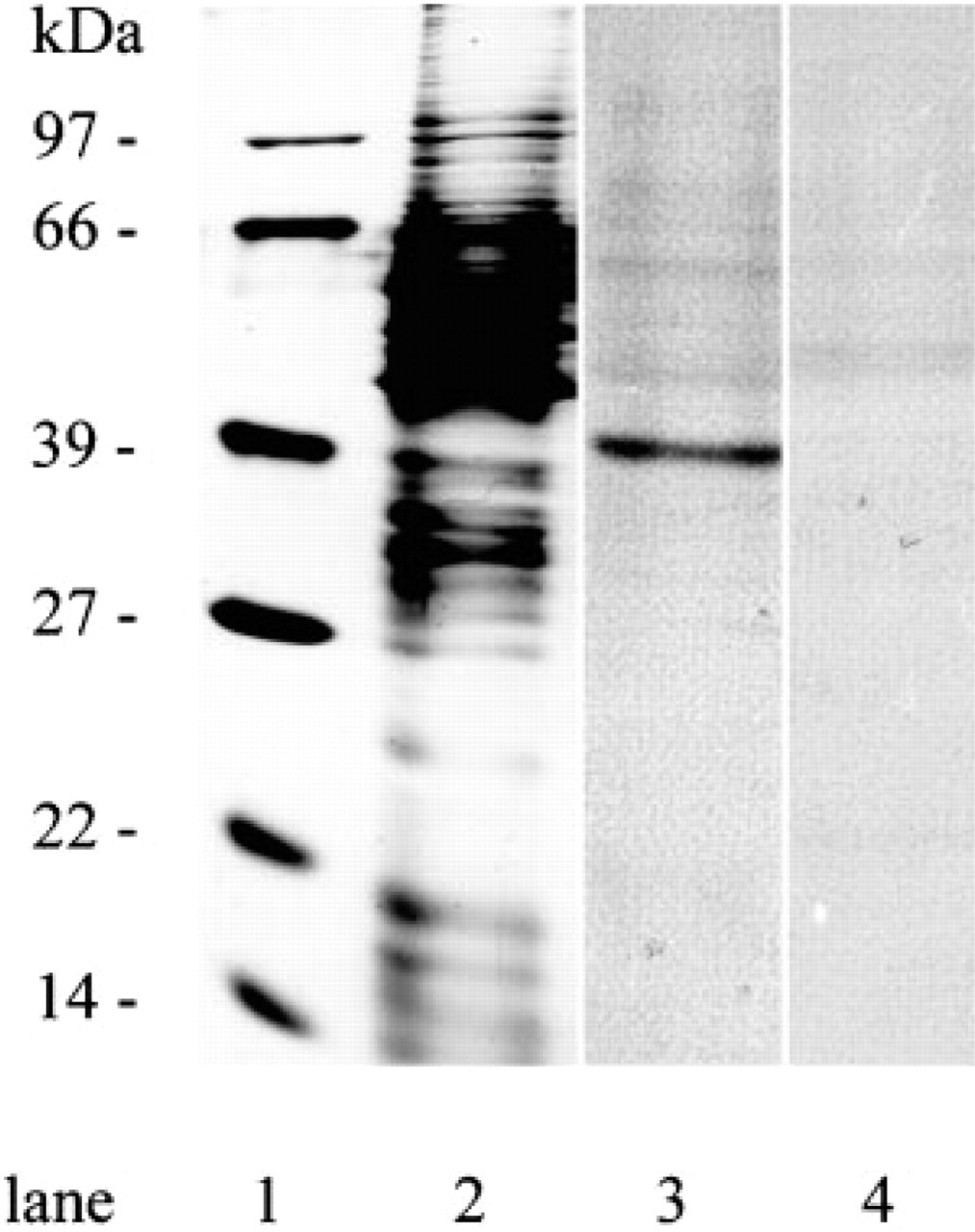
Western blotting of cultured microglial cell proteins with BCATm and BCATc antisera. Cell homogenate proteins (8 μg protein) were separated by SDS-PAGE, transferred to nitrocellulose, and Western blotting was performed as described in Materials and Methods. Lanes 1 and 2 show silver staining of the molecular mass marker proteins (Bio-Rad) and the microglial secondary culture cell proteins, respectively. Lanes 3 and 4 show immunostaining for BCATm and BCATc, respectively.
The second step in degradation of BCAA is oxidative decarboxylation of the transamination products, the BCKAs, which is catalyzed by the mitochondrial BCKD enzyme complex. This step commits the BCAA carbon skeleton to the degradation pathway. Therefore, the cellular distribution of this enzyme was investigated in cultured brain cells. Double staining of neuron-rich primary cultures with BCKD antiserum, which recognizes the E2 subunit of the BCKD, and with monoclonal antibodies against the neuronal marker GAP-43, revealed co-localization of the two proteins, indicating that neurons express BCKD (Figures 6A–6C). The fluorescence signal for BCKD was observed in the cell somata and also in the cell processes (Figure 6A). Because cultured neurons grow preferentially in cell clusters and the portion of cytoplasm around their cell nuclei is rather small, the characteristic punctate staining for the mitochondrial BCKD is difficult to see in neurons. In the experiment shown in Figures 6D–6E, an astroglia-rich primary culture was used to examine the presence of BCKD in astroglial cells. BCKD antiserum was applied in combination with an antibody against the astroglial cell marker GFAP. Staining for BCKD revealed co-localization of BCKD and GFAP in flat and extended cells of irregular shape. In contrast to the staining of neurons, in astroglial cells the punctate fluorescence signal for mitochondrial BCKD could be seen clearly in the extended cytoplasm of these cells (Figure 6D). The cell nuclei were not stained. In addition to GFAP-positive astroglial cells, other GFAP-negative cell types stained positive for BCKD (Figures 6D–6F). Most of these cells had small round somata carrying several thin and elongated processes characteristic of oligodendroglial cells and O2A lineage cells. In Figures 6G–6I, a microglial secondary culture was stained simultaneously for BCKD and for the microglial marker CR3. CR3-positive cells displayed intense fluorescence staining of punctate and tube-like structures, which were located in the cytosol. Again, the nuclei were not labeled. The cell periphery of the microglial processes was less intensely stained, most likely due to the lower occurrence of mitochondria. The results suggest that, in contrast to each of the BCAT isoenzymes, BCKD is expressed ubiquitously in rat brain cell cultures.
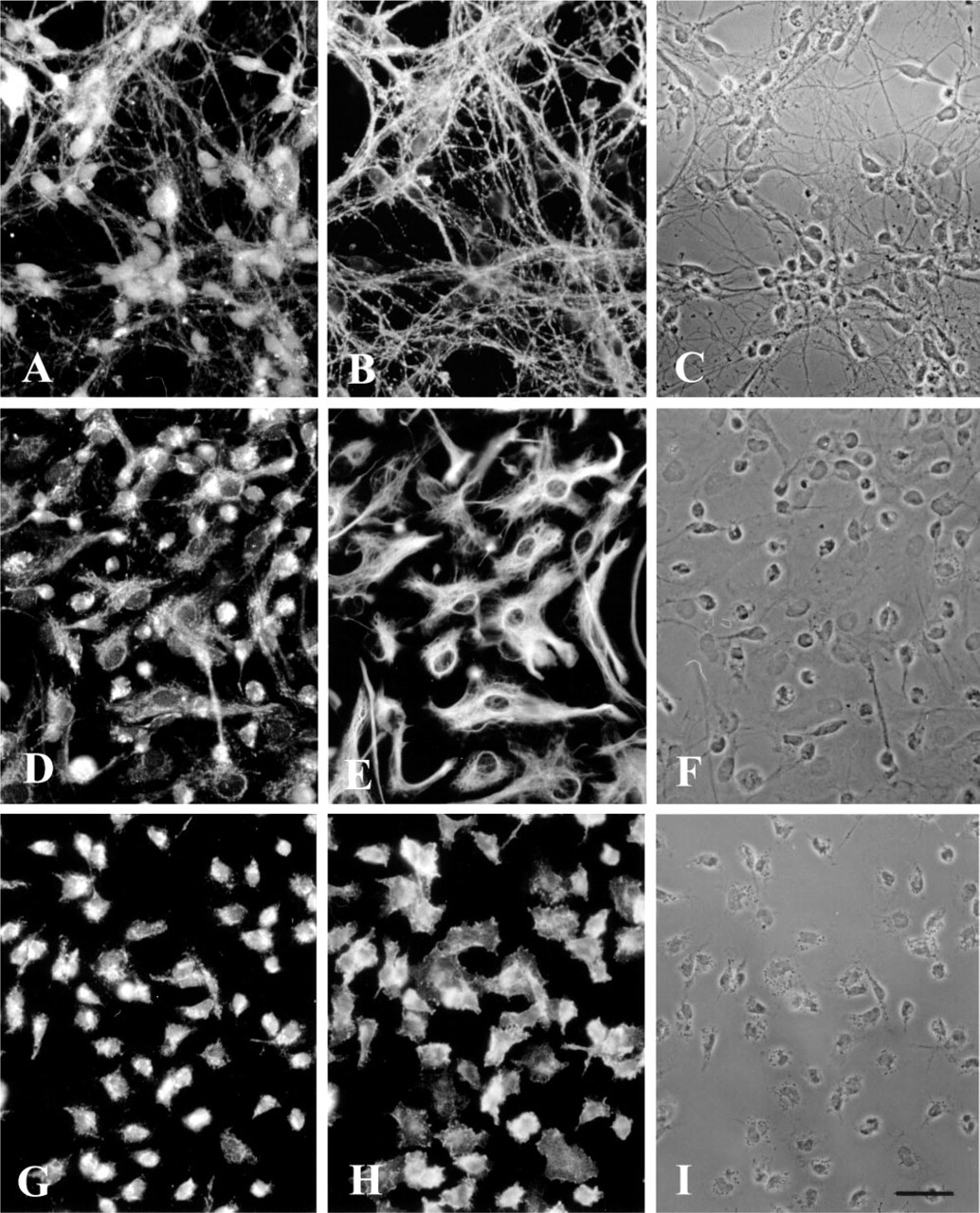
Immunofluorescence staining of a neuron-rich primary culture (
Discussion
This study shows that BCATc is the sole BCAT isoenzyme expressed in rat cortical neurons in culture, whereas BCATm is the isoenzyme form found in microglial cells in culture. In a separate immunocytochemical study, we found previously that BCATm is the predominant isoenzyme in cultured astroglial cells, while BCATc, but not BCATm, is found in high concentration in cultured oligodendroglial cells and O2A progenitors (Bixel et al. 1997). Therefore, the BCAT isoenzymes exhibit cell-specific expression in primary cultures of rat brain cells. In contrast, the E2 subunit of BCKD appears to be expressed in all cultured brain cells that have been examined (Figure 6). These results are consistent with measurements of BCAT isoenzyme and BCKD E2 subunit protein levels in primary astrocyte and neuronal cell cultures (Hutson et al. 1998).
Yudkoff and co-workers (1996a,b; Yudkoff 1997) and Bixel et al. (1996) have proposed that BCAAs play an important role in providing nitrogen for the glutamate/glutamine cycle. In the original glutamate/glutamine cycle (Benjamin and Quastel 1972; Berl et al. 1977; Hamberger et al. 1977), glutamate released into the synaptic cleft during neuronal activity is rapidly removed by nearby astrocytes (Hösli et al. 1986) and is converted to glutamine (Martinez–Hernandez et al. 1977). After export to juxtaposed glutamatergic neurons, the glutamine is used to regenerate the neuronal glutamate pool in a reaction catalyzed by glutaminase (Chaplin et al. 1976; Weiler et al. 1979; Shambaugh and Koehler 1981; Shank and Aprison 1981). The role of BCAAs in glutamate metabolism is based on studies showing that BCAAs can label a significant fraction of the glutamate nitrogen in brain cell cultures (Yudkoff et al. 1983, 1994) and in vivo (Kanamori et al. 1998). Evidence for the cycle comes from studies showing that leucine oxidation is slow in cultured astroglial cells (Bixel and Hamprecht 1995; Hutson et al. 1998) and that this results in release of the transamination product of leucine, α-ketoisocaproate, from the astrocytes (Bixel and Hamprecht 1995; Yudkoff et al. 1996a), and from investigations showing preferential transamination of α-ketoisocaproate in astrocytes, synaptosomes, and cultured neurons (Shambaugh and Koehler 1983; Yudkoff et al. 1996b; Hutson et al. 1998). In this nitrogen cycle, BCAA transamination in astrocytes donates nitrogen to glutamate (subsequently converted to glutamine) followed by release of BCKA by astrocytes. Reamination of the BCKA in neurons is followed by release of BCAAs. Uptake of BCAAs by astrocytes would result in transfer of nitrogen back to the astrocyte (Bixel et al. 1996; Yudkoff et al. 1996b; Yudkoff 1997).
The BCAA nitrogen shuttle has been expanded to include an obligatory role for these amino acids for optimal rates of de novo glutamate synthesis (Hutson et al. 1998). A significant fraction of the glutamate taken up by the astrocytes is converted to α-ketoglutarate and subsequently metabolized to lactate (Yu et al. 1983; Sonnewald et al. 1993; Hutson et al. 1998). The flow of citric acid cycle intermediates to glycolytic intermediates means that anaplerosis must take place at an equivalent rate to recover the lost carbon of glutamate, i.e., glutamate must be synthesized de novo. The anaplerotic enzyme pyruvate carboxylase is localized in astroglia (Shank et al. 1985; Cesar and Hamprecht 1995). Previous studies of the rate of anaplerosis in cultured astrocytes indicated that conversion of the excess citric acid cycle intermediates, formed by pyruvate carboxylase, to glutamate and glutamine could be severely limited by lack of a source of nitrogen (Yudkoff et al. 1996a, b; Gamberino et al. 1997; Hutson et al. 1998). Because NH3 could not supply the needed nitrogen (Gamberino et al. 1997), it was suggested that BCAAs might be the physiological source for the α-amino nitrogen of glutamate (Gamberino et al. 1997; Hutson et al. 1998).
The immunocytochemistry results presented here (Figures 2 and 3) and by Bixel et al. (1997) suggest that BCATm is the predominant isoenzyme in astrocytes. The predominance of BCATm in astroglial cultures and localization of BCATc in neurons suggest that BCATc is responsible for the production of BCAA in the neurons. This hypothesis is also consistent with the lack of effect of the BCATc-specific drug gabapentin on glutamate metabolism in astrocyte cultures and the significantly greater inhibition of BCAA metabolism by the drug in neuronal cultures (Hutson et al. 1998).
The BCAA nitrogen shuttle hypothesis makes no prediction about the location of the BCKD enzyme complex, but changes in activity of this enzyme could also affect operation of the cycle. Becausee the BCKD irreversibly decarboxylates the BCAA transamination products, the BCKAs, it is the committed step in BCAA oxidation and results in a net transfer of nitrogen to the cell in which this reaction occurs. The ubiquitous expression of BCKDs in rat brain cells in culture suggests that all types of brain cells can oxidize the BCAAs. Recently, we immunocytochemically localized β-methylcrotonyl-CoA carboxylase (β-MCC), an enzyme unique to the leucine catabolic pathway, in cultured astroglial and neuronal cells (Bixel and Hamprecht 2000). All astroglial cells were found to express β-MCC, and the majority of neurons also contained β-MCC. The expression of BCKD (present work) and β-MCC (Bixel and Hamprecht 2000) in neurons suggests that both cell types can degrade BCAAs. However, the predominant fate of BCAAs in neurons appears to be a release of amino acids for export of amino nitrogen to astrocytes (Yudkoff et al. 1996b).
BCAT isoenzymes also exhibit cell-specific expression in cultured rat brain cells other than astrocytes and neurons (Figure 4; Bixel et al. 1997). In the present study, microglial cells were found to express BCATm and BCKD. In a previous study we showed that BCATc was the sole BCAT isoenzyme expressed in oligodendrocytes and O2A progenitor cells (Bixel et al. 1997). The function(s) of BCAAs in these brain cells is not yet known, although one can speculate that the degradation products of BCAAs may contribute to lipid synthesis in oligodendrocytes. Future work will have to determine the cellular distribution of the isoenzymes in rat brain and the physiological functions of these aminotransferases in situ.
Footnotes
Acknowledgements
Supported by the NATO Collaborative Research Grants Program CRG 950864 and by grant DK 34738 (SMH) from the National Institutes of Health.
We thank Dr Brigitte Pfeiffer, Dr Heinrich Wiesinger, and Mr Oliver Kranich for kindly providing neuron-rich primary cultures, and Agus Suryawan for testing the specificity of the BCKD antiserum.