
Abstract
Expression of neutral glycosphingolipids (GSLs) and gangliosides in normal lymphoid tissues and cells has been studied mostly by biochemical and immunochemical analysis of lipid extracts separated by thin-layer chromatography. GSLs and gangliosides involved in the GM1b biosynthetic pathway were assigned to T-lymphocytes, whereas B-cell gangliosides and GSLs have been poorly characterized in former publications. We used specific polyclonal antibodies in immunohistochemistry and flow cytometry to analyze the distribution of globotriaosylceramide (Gb3Cer), globoside (Gb4Cer), gangliotriaosylceramide (Gg3Cer), gangliotetraosylceramide (Gg4Cer), and gangliosides GM3 and GalNAc-GM1b in the mouse thymus, spleen, and lymph node. Immature thymocytes expressed epitopes recognized by all antibodies, except for anti-Gb4Cer. Mature thymocytes bound only antibodies to GalNAc-GM1b, Gg4Cer, and Gb4Cer. In secondary lymphoid organs, antibodies to globo-series GSLs bound to vascular spaces of secondary lymphoid organs, whereas the ganglio-series GSL antibodies recognized lymphocyte-containing regions. In a Western blotting analysis, only Gal-NAc-GM1b antibody recognized a specific protein band in all three organs. Flow cytometric analysis of spleen and lymph node cells revealed that B-cells carried epitopes recognized by all antibodies, whereas the T-cell GSL repertoire was mostly oriented to ganglio-series-neutral GSLs and GM1b-type gangliosides. The results of immunohistochemistry and flow cytometry were not always identical, possibly because of crossreactivity to glycoprotein-linked oligosaccharides and/or differences between cell surface carbohydrate profiles of isolated cells and cells in a tissue environment.
Keywords
G
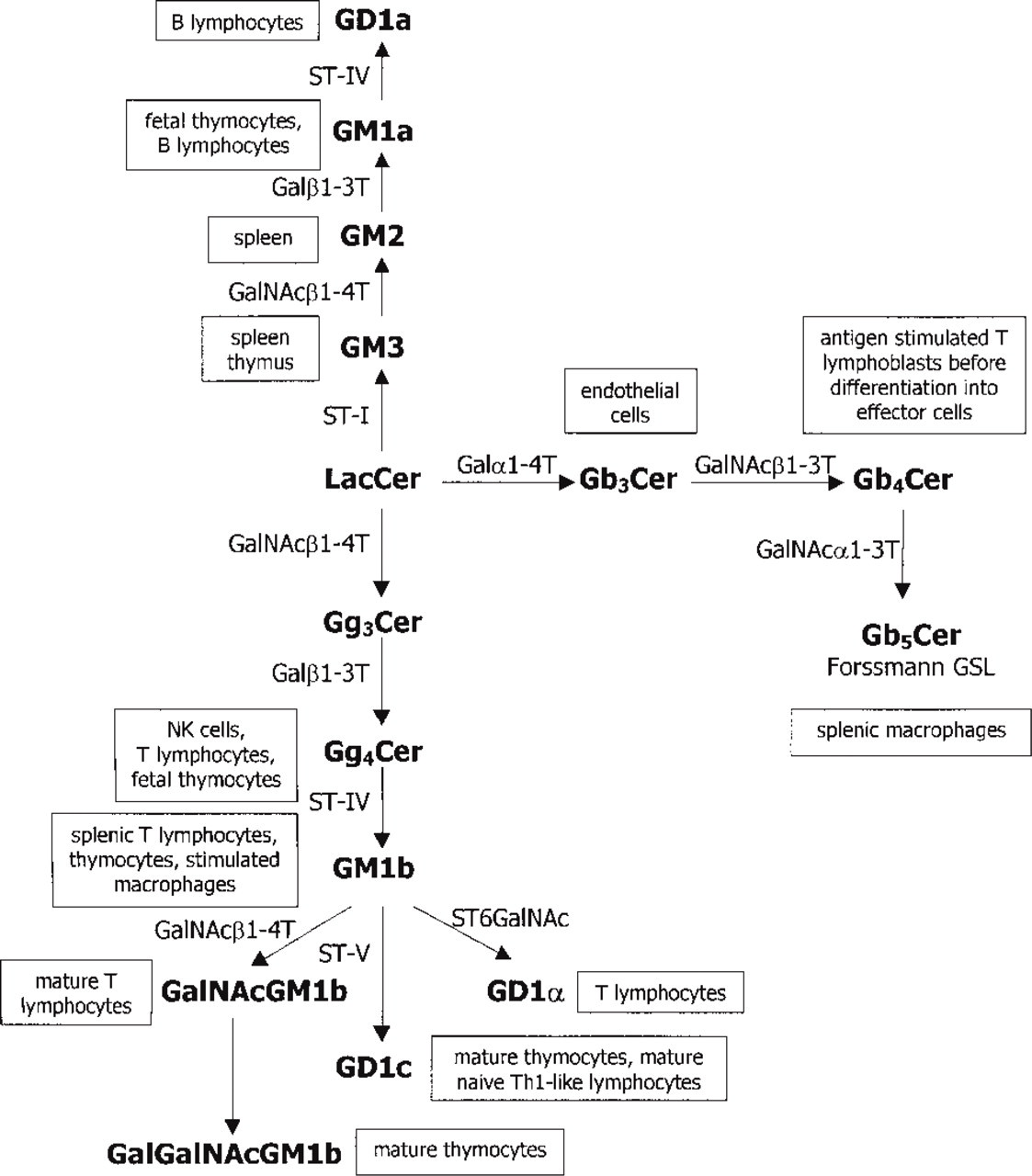
A schematic overview of glycosphingolipid biosynthesis and expression in murine lymphoid cells and tissues. Galα1–4T, galactosyl-(α1–4)-transferase; Galβ1–3T, galactosyl-(β1–3)-transferase; GalNAα1–3T, N-acetyl-galactosaminyl-(a1–3)-transferase; GalNAcβ1–3T, N-acetyl-galactosaminyl-(β1–3)-transferase; GalNAcβ1–4T, N-acetylgalactosaminyl-(β1–4)-transferase; ST-I, ST-IV and ST-V, sialyl-transferases I, IV, and V, respectively; ST6GalNAc, sialyltransferase that transferes sialic acid to position 6 of N-acetyl-galactosamine residue.
The GSL expression pattern changes with the cells' functional status and can be influenced by a variety of microenvironmental factors, including cytokines. Interferon-7 alters the expression of endothelial cell surface GSLs (Gillard et al. 1990), and constitutive expression of IL-3 gene by transfected cells leads to altered ganglioside expression (Tsunoda et al. 1995). The ganglioside phenotype of resting B-cells is altered by endotoxin stimulation (Pörtner et al. 1993) and the same is true for concanavalin A-stimulated T-cells (Horikawa et al. 1991).
Most of the above studies were performed by biochemical and structural analysis of GSL extracts from tissues or cultured cells, which may not reflect their expression in vivo. The GSL expression pattern is different in cultured cells vs those in vivo (Li et al. 1993). Studies of the histological distribution of GSL in normal lymphoid organs and lymphocyte populations in vivo are rare (Nakamura et al. 1995), as opposed to a number of studies on malignant cells (Zhang et al. 1997; Chammas et al. 1999). To extend our previous biochemical studies of GSLs in lymphoid organs (Müthing et al. 1989; Müthing 1997; Markotić et al. 1999), we used antibodies developed for biochemical analysis of GSLs to detect them in cryosections of normal lymphoid organs and in flow cytometry on cells belonging to the B- or T-lymphocyte population. We used specific antibodies to Gg3Cer and Gg4Cer, which are precursors in the biosynthesis of GM1b-type gangliosides, and to GalNAc-GM1b as a representative of GM1b-type gangliosides. Anti-Gb3Cer and anti-Gb4Cer antibodies were used to detect distribution of globoseries neutral GSLs, which are also expressed on lymphoid cells (Mühlradt et al. 1984; Mangeney et al. 1991). Antibody against GM3 was used because this structure is the precursor on a-, b-, and c-pathways of ganglioside biosynthesis (van Echten and Sandhoff 1993) and is widely distributed in various mouse organs, including lymphoid tissues (Nakamura et al. 1988).
In this study we used a combination of immunohistochemistry, flow cytometry, and Western blotting analysis to obtain information about GSL expression in lymphoid cells and tissues.
Materials and Methods
Antibodies
Chicken polyclonal antibodies were used to detect Gg3Cer, Gb3Cer, Gb4Cer, GM3, and GalNAc-GM1b, and a rabbit polyclonal antibody to detect Gg4Cer (Table 1). Chicken antibodies were of the IgY isotype, the equivalent of IgG in mammals. All antibodies were produced and characterized by the laboratory of Dr. J. Müthing and were used for thinlayer chromatography (TLC) immunostaining of separated gangliosides and neutral GSLs (see references in Table 1). Dr. Müthing can be contacted for all information on antibody availability. Polyclonal chicken anti-Gg3Cer antibody was produced with HPLC-purified Gg3Cer according to the method of Kasai et al. (1980). The antibody specifically binds to the GalNAcβ4Gal terminus and does not crossreact with globo- or neolacto-series GSLs (our unpublished data). Preimmune chicken or rabbit sera were used as negative controls in all experiments. The specificity of the antibodies was additionally confirmed by flow cytometric competition assays using excess purified glycolipids. Individual glycolipids effectively inhibited binding of the relevant antibody but did not affect binding of antibodies specific for other glycolipids (data not shown).
Secondary alkaline phosphatase-conjugated affinity chromatography-purified rabbit anti-chicken and goat anti-rabbit antibodies were used for immunohistochemistry, and dichlorotriazinyl-amino fluorescein (DTAF)-conjugated antibodies (Dianova; Hamburg, Germany) for flow cytometry.
A monoclonal anti-mouse CD19 antibody, specific for the B-cell surface marker expressed by all mature B-cells, and a monoclonal anti-mouse CD3∊ antibody, specific for the T-cell receptor-associated molecule and expressed on all mature T-cells, both conjugated with phycoerythrin (PE), were used for double flow cytometric staining (Pharmingen; San Diego, CA).
Polyclonal antibodies used for immunohistochemical and flow cytometric detection of GSLs in mouse lymphoid organs
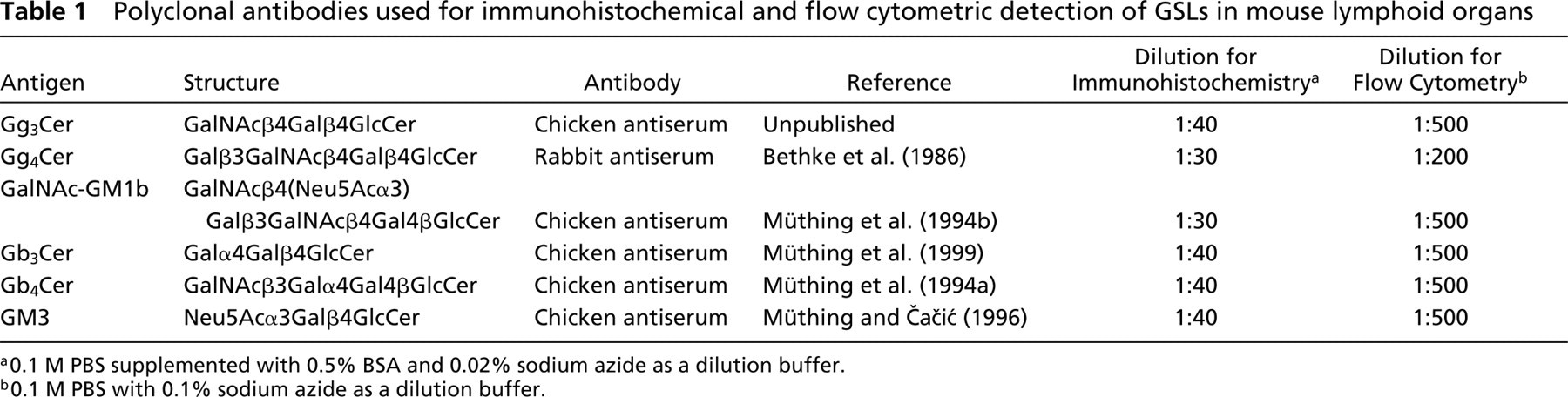
a0.1 M PBS supplemented with 0.5% BSA and 0.02% sodium azide as a dilution buffer.
b0.1 M PBS with 0.1% sodium azide as a dilution buffer.
Animals
Female C57BL/6 mice, 6−8 weeks of age, were used in all experiments. For immunohistochemistry, animals were perfused with 20 ml of cold (4C) 0.1 M PBS, pH 7.4, immediately after sacrifice by CO2 anesthesia, and then with 20 ml of 4% formaldehyde in 0.1 M PBS, pH 7.4. After perfusion, whole organs were removed and postfixed for 2 hr in cold (4C) 2− formaldehyde in 0.1 M PBS, pH 7.4. Tissue was then rinsed in PBS, chilled in isopentane (−80C) (Merck; Darmstadt, Germany), and stored at −80C. For flow cytometry, whole organs were excised in ice-cold 0.1 M PBS with 0.1% NaN3 and were homogenized to obtain a single-cell suspension. Erythrocytes, especially abundant in spleen homogenates, were removed using a lysis buffer (150 mM NH4Cl, 1 mM KHCO3, and 0.1 mM Na2EDTA, pH 7.4).
Immunohistochemistry
Tissue sections 5 μm thick were cut on a cryomicrotome (Leica; Nussloch, Germany) at −20C, mounted on gelatinprecoated glass slides, and air-dried for 2 hr. All subsequent steps were performed at room temperature. Nonspecific antibody binding was blocked by incubating the sections with 1% bovine serum albumin (BSA) in 0.1 M PBS, pH 7.4, for 1 hr. Blocking solution was poured off and each section was incubated for 2 hr with 30 μl of polyclonal primary antibody (Table 1), diluted with 0.1 M PBS supplemented with 0.5% BSA and 0.02% NaN3. Preimmune serum, derived from the same animal species in which antibody was raised, was used as a negative control at the same dilution as the primary antibody. Sections were then rinsed three times in 0.1 M PBS and incubated for the next 2 hr with alkaline phosphatase-conjugated secondary antibody diluted 1:500 using the same buffer as for the primary antibody dilution, followed by three washes in 0.1 M PBS. Visualization of antibody binding was achieved with naphthol-AS-MX phosphate and Fast Red substrate (Sigma; St Louis, MO), followed by hematoxylin counterstaining. Sections were mounted with Mowiol (Hoechst; Frankfurt, Germany) as previously described (Čaĉić et al. 1994).
Antibody staining was evaluated under a standard light microscope and staining intensity was graded as – for no staining, + for weakly positive staining, ++ for moderate staining, and + + + for very intensive staining.
To confirm the lipid nature of antibody binding structures, sections were pretreated with methanol and then with chloroform/methanol (1:1, v/v), each for 10 min, before immunostaining (Čačić et al. 1995). This procedure should extract lipids from cryosections and the residual binding indicates the presence of protein-linked oligosaccharides resembling side chains of GSLs. After lipid extraction, sections were air-dried and stained according to the same procedure as described for non-extracted sections.
Flow Cytometry
Single-cell suspensions of spleens, thymi, and lymph nodes were prepared in ice-cold 0.1 M PBS with 0.1 % sodium azide. After centrifugation, 106 cells were incubated with primary anti-GSL antibodies (Table 1) and/or 1 μg of PE-conjugated antibodies reactive to mouse CD19 or CD3∊ (Pharmingen) for 30 min on ice. Antibodies were diluted in 0.1 M PBS with 0.1% NaN3. After two washes in 0.1 M PBS with 0.1% sodium azide, 0.5 μg of secondary DTAF-conjugated, affinity chromatography-purified rabbit anti-chicken IgY and goat anti-rabbit IgG antibodies (Dianova) was added and incubated on ice for the next 30 min. Finally, cells were resuspended in 1 ml of 0.1 M PBS with 0.1% sodium azide.
Two-color fluorescence was measured at the excitation wavelength of 496 nm, using a FACSCalibur (Becton–Dickinson; San Jose, CA). Fluorescence was further quantified on the population of lymphocytes gated according to FSC (forward scatter, proportional to cell size) vs SSC (side scatter, proportional to cell complexity) dot-plots. A total of 104 cells was analyzed. Negative controls for anti-CD19 and anti-CD3 antibodies were non-immune species-matched, PE labeled immunoglobulins. Negative controls for anti-GSL antibodies were preimmune sera derived from the same species in which antibody was raised. They were used in the same dilution as the primary antibody, followed by the incubation with DTAF-labeled secondary antibody. Nonspecific binding of secondary antibody was excluded by incubating the cells only with the DTAF-labeled secondary antibody.
Western Blotting Analysis
Thymi, spleens, and lymph nodes were dissected out from their fibrous capsules and homogenized in ice-cold 20 mM imidazole, 250 mM sucrose, pH 7.38, with the addition of protease inhibitor (No. 1873580; Boehringer, Mannheim, Germany). The homogenate was centrifuged at 1500 rpm and 4C for 15 min. Supernatant was removed into a clean tube and centrifuged for another 10 min at 14,000 rpm at 4C. Protein concentration was determined using a commercial kit (No. 500-0001; Bio Rad, Vienna, Austria). Samples were then mixed with sample buffer (Bio Rad) containing 2% SDS, and heated at 100C for 10 min. Equal amounts of proteins (20 μg) were separated on a 12% polyacrylamide gel and electrophoretically transferred onto a nitrocellulose membrane. After blocking with 3% BSA in Tris-buffered saline (TBS; 10 mM Tris-HCl, 150 mM NaCl, pH 7.4), membranes were incubated with anti-GSL primary antibody diluted in TBS 1:500. After four washes in TBS with 0.02% Tween-20, the membranes were incubated with affinity chromatography-purified alkaline phosphatase-conjugated rabbit anti-chicken or goat anti-rabbit secondary antibody, diluted 1:100 in TBS. Membranes were then washed three times in 0.02% Tween-20 in TBS and once in TBS. Finally, proteins were visualized using a 5-bromo-4-chloro-3-indolyl phosphate and nitroblue tetrazolium (BCIP/NBT) developing system (Kirkegaard & Perry Laboratories; Gaithersburg, MD) according to the manufacturer's instructions. A low molecular weight electrophoresis calibration kit was obtained from Amersham Pharmacia (New York, NY).
Results
Biochemical analysis of lymphoid tissues from C57BL/6 mice showed that GSL fractions isolated and purified from thymus, spleen, and lymph nodes contain GM1b, GalNAc-GM1b, and GM3 gangliosides, as well as Gb3Cer, Gb4Cer, Gg3Cer, and Gg4Cer neutral GSLs (Markotić et al, 1999; and our unpublished results). To obtain optimal antibody binding to tissue sections, we tried different fixation methods (Schwarz and Futerman 1997), including staining of unfixed cryosections. For antibodies used in this study, perfusion with 4% paraformaldehyde and prefixation with 2% paraformaldehyde before tissue freezing and sectioning gave the best results. The antibody staining after paraformaldehyde prefixation was slightly less intense than that on unfixed cryosections but gave much better visualization of cellular compartments in lymphoid organs (data not shown). Control preimmune sera did not show any background binding at dilutions used in histochemistry or flow cytometry.
Tables 2–4 summarize the data on GSL expression in the thymus, spleen, and lymph nodes obtained by immunohistochemistry and flow cytometry. The data are from a representative experiment from a series of three experiments with similar results.
Thymus
Histologically, the thymus consists of a cortical part harboring immature thymocytes, closely associated with specialized cortical epithelium (Janeway and Travers 1996). During the maturation process, cells move towards the medullar space containing mature thymocytes in the network of medullar epithelial cells and dendritic cells, and leave the thymus to migrate to secondary lymphoid organs. Subcapsular cortical thymocytes do not express CD3∊; deeper cortical cells are immature thymocytes going through T-cell receptor gene rearrangement, resulting in a low expression of CD3∊. Mature T-cells that have completely rearranged their T-cell receptor gene are located in the medulla and express high levels of the CD3∊ molecule.
Strong binding of GalNAc-GM1b antibody was localized in the medullar region of the thymus (Figure 2A, upper panels) and was mostly of lipid nature, with trace positivity still visible in the perivascular spaces after pretreatment with organic solvents (Figure 2A, lower panels). Weak binding that was not affected by lipid extraction also persisted in the thymic cortex.
Because a TLC immuno-overlay detected rather small amounts of GalNAc-GM1b in the thymus of C57BL/6 mice (Markotić et al. 1999), we performed Western blotting analysis of cell homogenates of thymus, spleen, and lymph node to assess eventual binding of the antibody to glycoproteins. A specific band migrating above the 43-kD protein marker was detected by anti-GalNAc-GM1b antibody in Western blotting analysis of protein extracts from all three lymphoid organs (Figure 3), whereas other glycolipid antibodies did not show specific binding (data not shown).
Immunohistochemical and flow cytometric analysis of GSL expression in the thymus

a–, no staining; +, weak positive staining; ++, moderate intensity staining; +++, strong staining.
bExpressed as percentage of lymphocyte population gated according to forward scatter vs side scatter dot-plots. The percentage of GSL single stain was similar to the sum of GSL staining on CD3 or CD19 positive cells, indicating that double labeling did not affect the binding of individual antibodies.
cThe proportion of single-stained CD3+high mature T-lymphocytes was 5–15%.
Immunohistochemical and flow cytometric analysis of GSL expression in the spleen
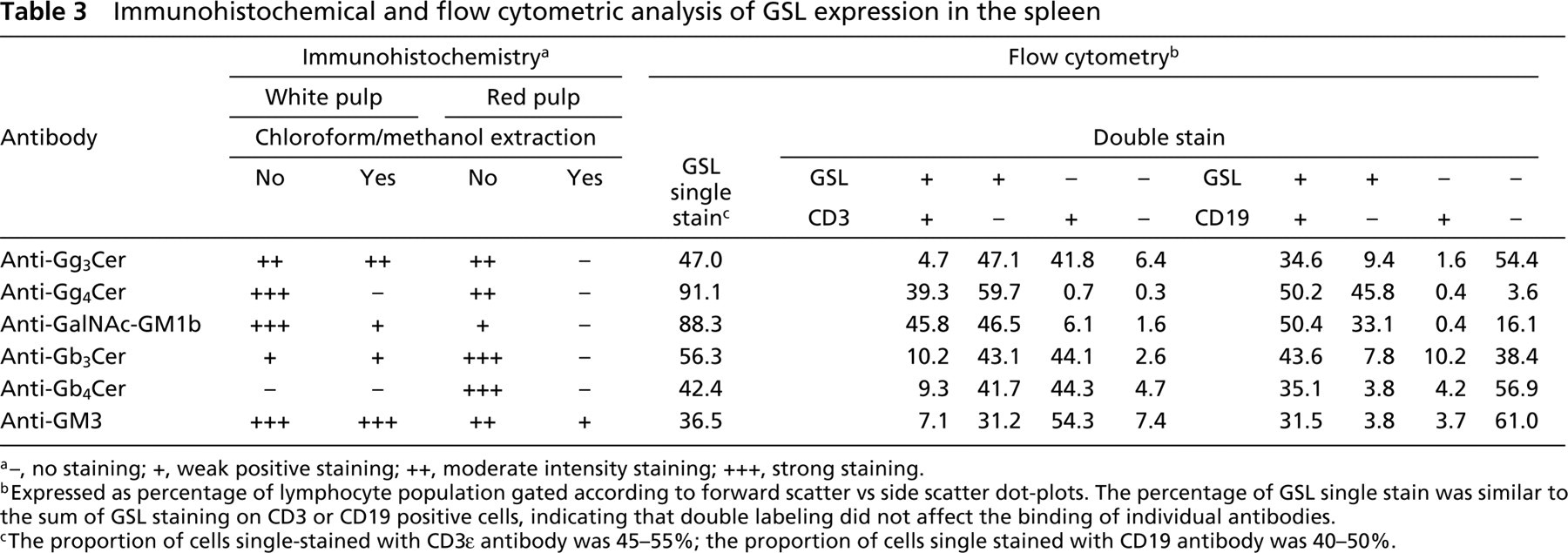
a–, no staining; +, weak positive staining; ++, moderate intensity staining; +++, strong staining.
bExpressed as percentage of lymphocyte population gated according to forward scatter vs side scatter dot-plots. The percentage of GSL single stain was similar to the sum of GSL staining on CD3 or CD19 positive cells, indicating that double labeling did not affect the binding of individual antibodies.
cThe proportion of cells single-stained with CD3∊ antibody was 45–55%; the proportion of cells single stained with CD19 antibody was 40–50%.
Flow cytometry showed that the anti-GalNAc-GM1b binding structure was expressed on the surface of almost all thymocytes (Figure 4A), including mature CD3+high thymocytes (Figure 4B).
Anti-Gg4Cer antibody stained both the thymic cortex and medulla (Figure 2B, upper panels). Lipid extraction confirmed a completely lipid nature of the bound antigen in the thymic cortex, whereas weak binding remained in the thymic medulla (Figure 2B, lower panels). Flow cytometry revealed that about a third of thymocytes expressed Gg4Cer on their surfaces. These cells belonged to both mature CD3+high and immature CD3+low thymocytes. The majority of CD3– immature thymocytes did not express Gg4Cer on the cell surface (Figure 4B; Table 2).
Anti-Gb3Cer antibody strongly stained the cortex (Figure 2C, upper panels), and this staining was not altered by chloroform/methanol extraction. In contrast, intensely stained positive patches in the medulla were almost completely removed by lipid extraction, leaving only weak and diffuse perivascular staining (Figure 2C, lower panels). In flow cytometry, more than 80% of thymocytes bound anti-Gb3Cer (Figure 4A). Cells that did not bind anti-Gb3Cer were mature CD3+high thymocytes (Figure 4B; Table 2).
The histological distribution of anti-Gg3Cer antibody binding was similar to that of anti-Gg4Cer antibody, although more intense. Lipid extraction removed most of the medullar staining, except for the perivascular spaces, but did not affect staining of the cortex (Table 2). In flow cytometry, more than 80% thymocytes bound anti-Gg3Cer antibody (Figure 4A). Double staining revealed that those cells were immature CD3– or CD3+low thymocytes (Figure 4B). Mature CD3+high thymocytes did not bind anti-Gg3Cer antibody (Table 2).
Immunohistochemical and flow cytometric analysis of GSL expression in the lymph node
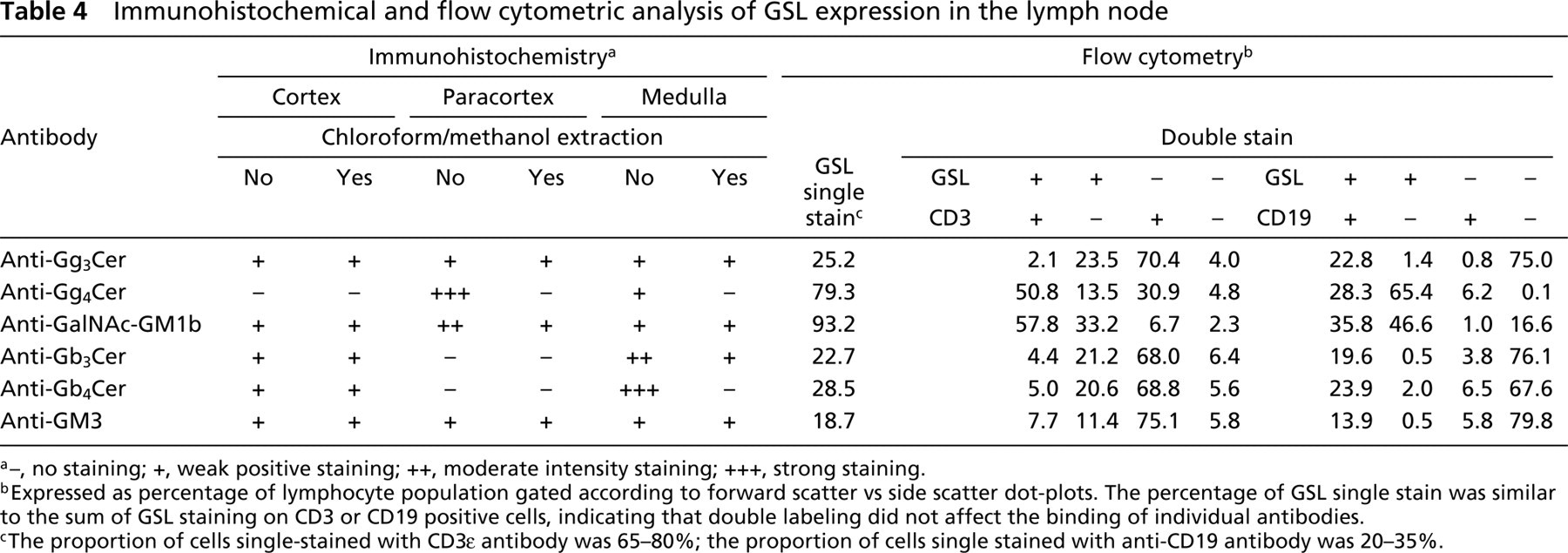
a–, no staining; +, weak positive staining; ++, moderate intensity staining; +++, strong staining.
bExpressed as percentage of lymphocyte population gated according to forward scatter vs side scatter dot-plots. The percentage of GSL single stain was similar to the sum of GSL staining on CD3 or CD19 positive cells, indicating that double labeling did not affect the binding of individual antibodies.
cThe proportion of cells single-stained with CD3∊ antibody was 65–80%; the proportion of cells single stained with anti-CD19 antibody was 20–35%.
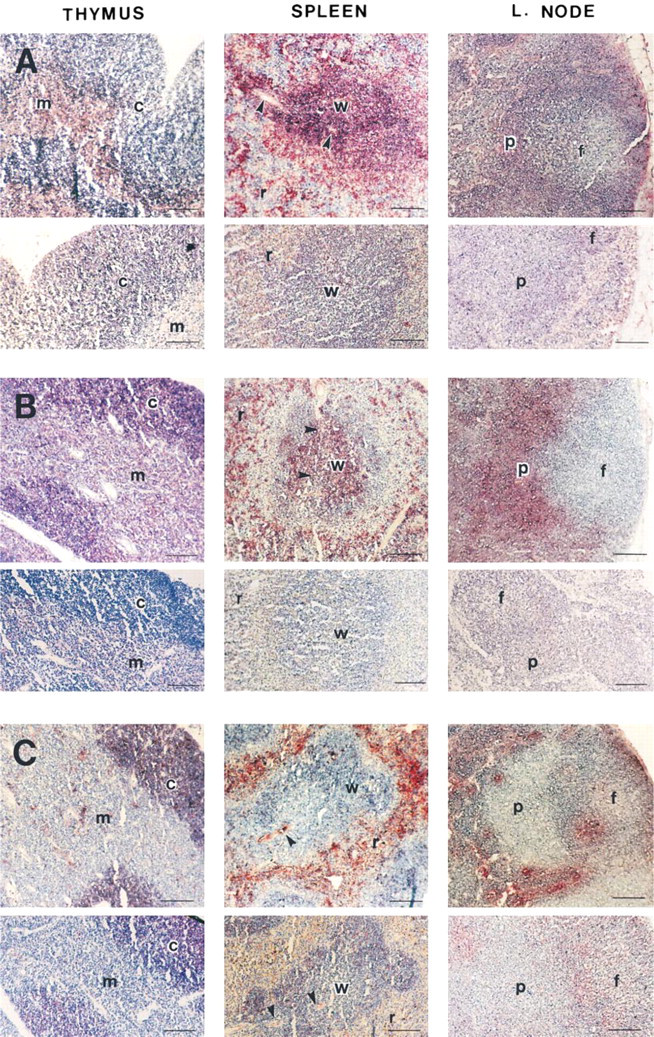
Light micrographs of thymus, spleen, and lymph node cryosections (original magnification ×100) immunostained with anti-GalNAc-GM1b (
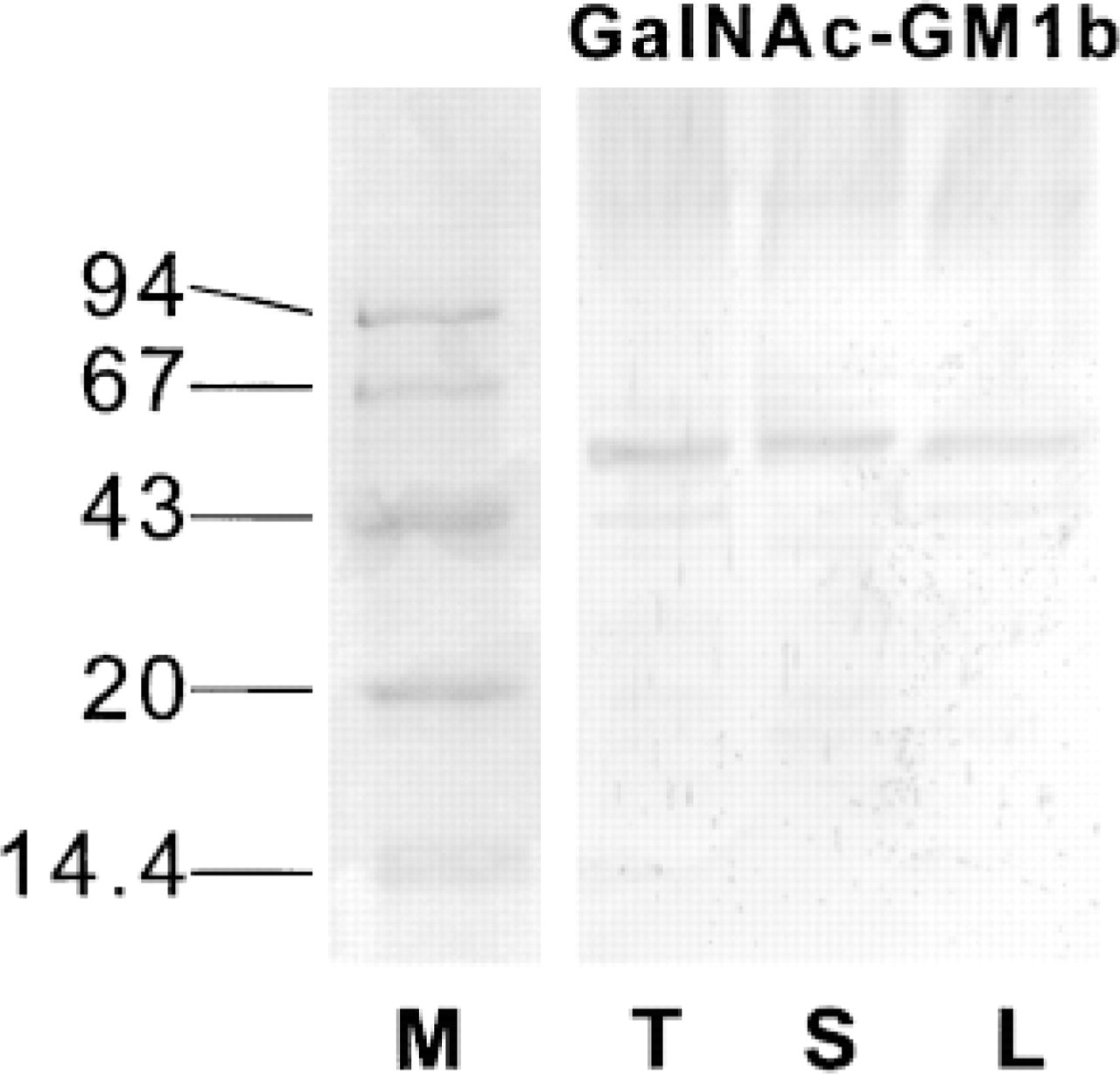
Western blotting analysis of protein extracts from the thymus, spleen, and lymph nodes of C57BL/6 mice. Membrane was immunostained with anti-GalNAc-GM1b antibody; molecular weight marker was stained with Coomassie blue. GalNAc-GM1b, staining with antibody to GalNAc-GM1b; M, molecular weight markers in kD; L, lymph node; S, spleen; T, thymus.
Anti-Gb4Cer antibody immunohistochemically labeled mostly perivascular spaces in the thymus, with the most prominent staining around blood vessels in the corticomedullar junction. The staining was readily removed with chloroform/methanol pretreatment (Table 2). In flow cytometry, 5.1% of thymocytes bound anti-Gb4Cer antibody (Figure 4A), those mostly belonging to the mature CD3+high population (Figure 4B).
Immunohistochemistry with anti-GM3 antibody yielded a similar staining pattern as the anti-Gb3Cer antibody, although somewhat more intense (Table 2). Flow cytometry revealed that half of the thymocytes bound anti-GM3 antibody (Table 2). These were mostly immature CD3– or CD3+low thymocytes (Figure 4A), because mature CD3+high thymocytes did not show anti-GM3 binding structures (Figure 4B).
Spleen
Spleen tissue consists of the white pulp, which harbors lymphocytes, and the red pulp, which is the site of erythrocyte destruction (Janeway and Travers 1996). The marginal sinus, bordered by the marginal zone of lymphocytes, encircles the white pulp. Central areas of the white pulp, around the central arteriole (periarteriolar sheaths) and within the marginal sinus, contain mainly T-lymphocytes, whereas the peripheral parts of the white pulp contain B-lymphocytes (Janeway and Travers 1996).
Anti-GalNAc-GM1b antibody bound to the white pulp of the spleen (Figure 2A, upper panels), without specificity for T- or B-cell-dependent zones, and to the marginal zone. It also stained the red pulp, but less intensely than other anti-GSL antibodies. Staining of the T-cell-dependent periarteriolar sheaths was completely abolished by lipid extraction, but some staining still remained in B-cell-dependent peripheral regions (Figure 2A, lower panels). Western blotting analysis revealed a band just above the 43-kD protein marker (Figure 3). In accordance with intensive immunohistochemical staining of the white pulp, antibody bound to almost 90% of splenocytes in flow cytometry (Figure 5A). A small proportion of cells that did not react with GalNAc-GM1b antibody were exclusively CD3+ T cells (Figure 5B). The entire CD19+ B-cell population was stained by GalNAc-GM1b antibody (Figure 5C).
Gg4Cer antibody staining was most intense in central periarteriolar sheaths of the white pulp, a T-cell-dependent zone (Figure 2B, upper panels). The periphery of the white pulp showed some positive staining and the red pulp was intensely stained. Lipid extraction completely abolished staining with Gg4Cer antibody (Figure 2B, lower panels). Anti-Gg4Cer antibody bound to more than 90% of all splenocytes (Figure 5A), and with even distribution between T- and B-lymphocyte populations of the spleen (Figure 5B and 5C, respectively; Table 3).
Gb3Cer antibody strongly bound to the spleen red pulp (Figure 2C, upper panels) and less intensely stained B-cell-dependent peripheral white pulp regions. T-cell-dependent zones of the white pulp were completely unstained except for a narrow acellular region around the central arteriole. Chloroform/methanol extraction completely abolished the positive staining of the red pulp but not of the B-cell-dependent periphery of the white pulp (Figure 2C, lower panels). Flow cytometry revealed that more than half of all splenocytes bound anti-Gb3Cer antibody (Figure 5A). These were mostly CD19+ B-cells (Figure 5C), whereas only a minor proportion of CD3+ T-cells bound anti-Gb3Cer (Figure 5B; Table 3).
Anti-Gg3Cer antibody intensely stained the B-cell-dependent peripheral parts of the white pulp, whereas the central periarteriolar sheath and the marginal sinus were not stained (Table 3). Staining of the white pulp was not altered by lipid extraction. The red pulp was also intensely stained with anti-Gg3Cer antibody, but this positivity was completely removed by lipid extraction. In flow cytometry, about half of all splenocytes were anti-Gg3Cer-positive (Figure 5A). These were mostly CD19+ B-cells (Figure 5C), whereas less than 5% Gg3Cer-positive cells were CD3+ T-cells (Figure 5B; Table 3).
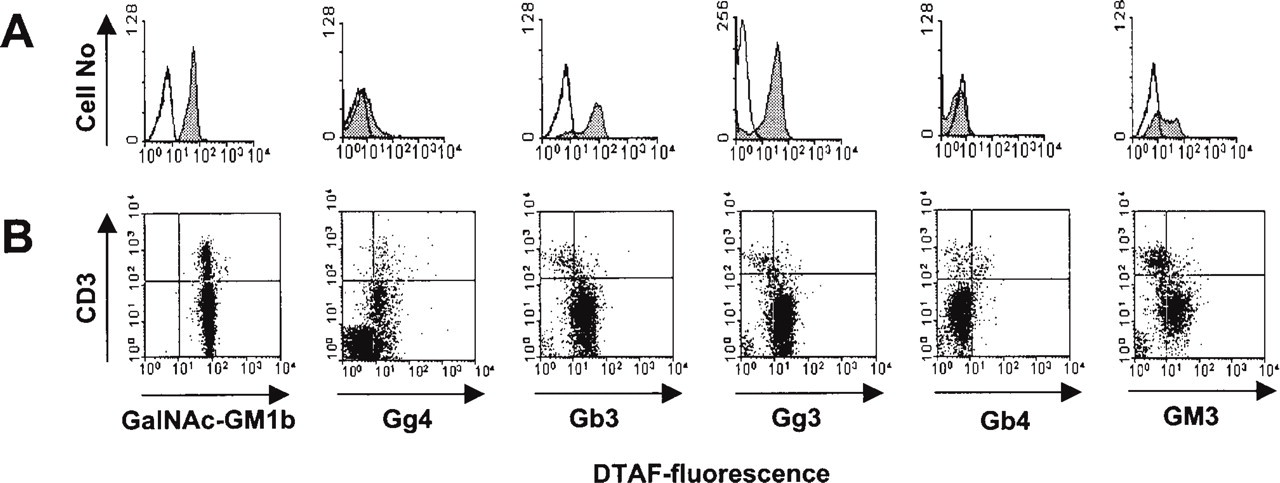
Flow cytometric analysis of the GSL expression on mouse thymocytes. (
Anti-Gb4Cer antibody reacted exclusively with the spleen red pulp and the narrow periarteriolar area, whereas the white pulp remained unstained (Table 3). Anti-Gb4Cer binding was abolished by chloroform/methanol pretreatment of the sections. Although the white pulp was completely unstained on spleen sections, the antibody bound to about 40% of the splenocytes in flow cytometry (Figure 5A), mostly to CD19+ B-cells (Figure 5C).
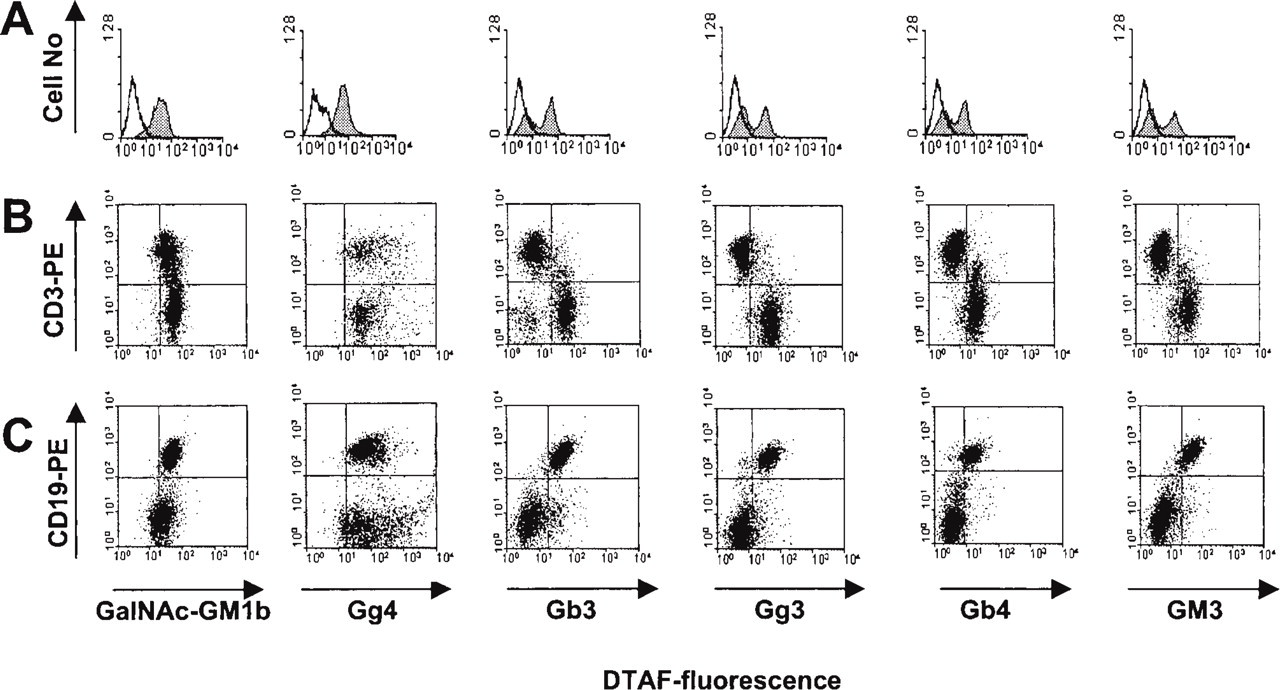
Flow cytometric analysis of GSL expression on mouse splenocytes. (
Anti-GM3 antibody intensely stained peripheral B- cell-dependent areas of the white pulp and the marginal sinus, but not the central periarteriolar T-cell-dependent zones (Table 3). White pulp positivity was resistant to lipid extraction and the red pulp positivity could be only partially abolished by this treatment. In accordance with the immunohistochemical findings, flow cytometry showed that anti-GM3 antibody bound almost exclusively to B-cells (Figure 5C) but only to a small percentage of T-cells (Figure 5B).
Lymph Node
Histologically, the lymph node consists of an outer cortex, which contains lymphoid follicles composed mostly of B-cells (Janeway and Travers 1996). T-cells are located in the deeper paracortical zone. The medullar region consists of medullary cords, containing plasma cells and macrophages, and a medullar sinus (Janeway and Travers 1996).
GalNAc-GM1b antibody weakly stained B-cell follicles in the cortical region and, somewhat more intensely, the T-cell-dependent paracortical area (Figure 2A, upper panels). Staining was not affected by lipid extraction (Figure 2A, lower panels). Flow cytometry was in accordance with such a diffuse staining pattern. Anti-GalNAc-GM1b antibody bound to more than 90% of lymph node lymphocytes (Figure 6A). The majority of T-cells and all lymph node B-cells expressed GalNAc-GM1b antibody binding epitope (Figure 6B and 6C).
By immunohistochemistry, anti-Gg4Cer antibody stained almost exclusively the T-cell-dependent paracortical region, with less intense staining in the medullar region (Figure 2B, upper panels). Lipid extraction completely abolished the staining (Figure 2B, lower panels). Flow cytometry showed positive binding to 80% of the lymph node lymphocytes (Figure 6A). Not only the CD3+ T-cells but also CD19+ B-cells were stained (Figure 6B and 6C; Table 4), although the B-cell-dependent cortical region was unreactive in immunohistochemistry.
The most intense staining with anti-Gb3Cer antibody was observed in the medullar area and cortical vessels (Figure 2C, upper panels). Trace positivity was observed in the cortical region. Chloroform/methanol pretreatment partially diminished anti-Gb3Cer antibody binding (Figure 2C, lower panels). Flow cytometry revealed that about 20% of all lymph node lymphocytes bound the antibody (Figure 6A). They were mostly CD19+ B-cells (Figure 6C), whereas the majority of CD3+ T-cells were unreactive (Figure 6B; Table 4).
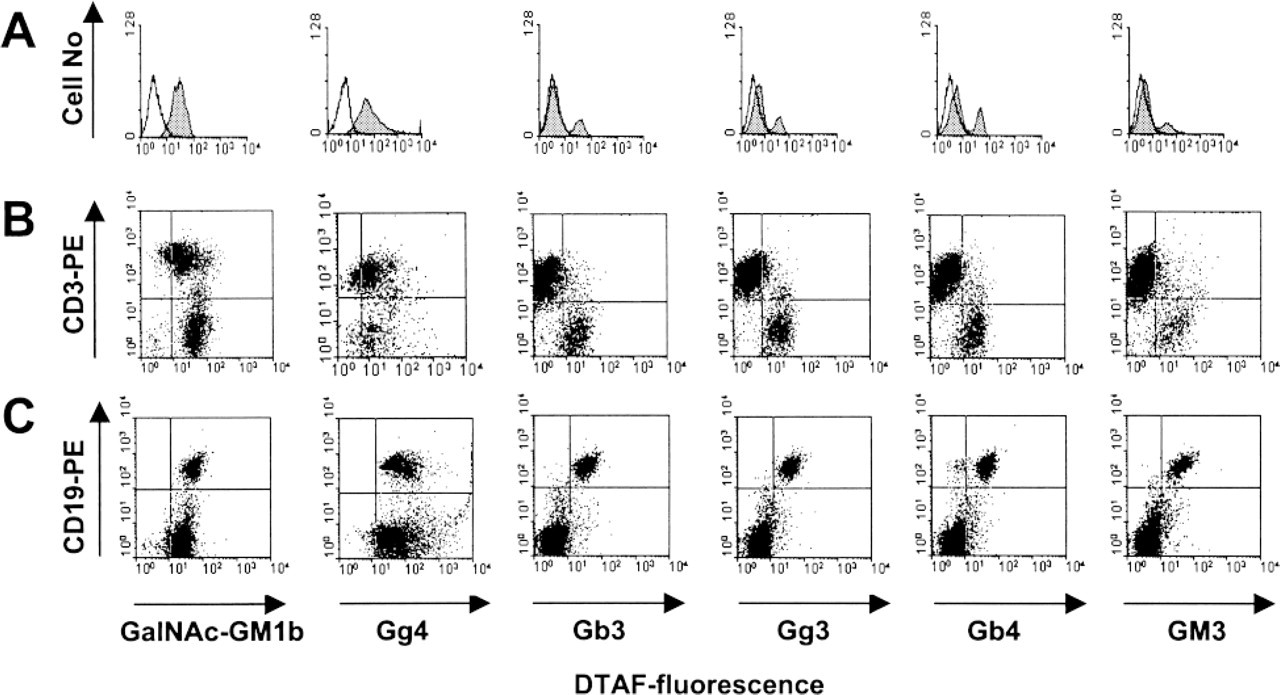
Flow cytometric analysis of GSL expression on mouse lymph node cells. (
The staining with anti-Gg3Cer antibody was diffuse and weak (Table 4). The staining was more intense in B-cell follicles of the cortical region, whereas a trace positivity was present in the paracortical and medullar areas. Lipid extraction decreased but did not abolish the staining (Table 4). In flow cytometry, 25% of all lymph node lymphocytes bound the anti-Gg3Cer antibody (Figure 6A). Most of them were CD19+ B-cells (Figure 6C and 6B; Table 4).
Staining with anti-Gb4Cer antibody was present only in the medullar area and cortical blood and/or lymph vessels, and chloroform/methanol pretreatment abolished the staining (Table 4). Flow cytometry revealed that about 30% of all lymph node lymphocytes bound anti-Gb4Cer antibody (Figure 6A). All CD19+ B-cells and a small fraction of CD3+ T-cells bound the antibody (Figure 6B and 6C; Table 4).
Anti-GM3 immunohistochemistry was similar to that of anti-Gg3Cer (Table 4), with dispersed, moderately intense staining in all lymph node regions, and was unaltered by lipid extraction. Flow cytometry revealed that the anti-GM3 antibody predominantly bound to CD19+ B-cells (Figures 6A–6C; Table 4).
Discussion
This study provides a comprehensive analysis of tissue and cellular expression of GSLs in lymphoid organs in vivo, using well-characterized anti-GSL polyclonal antibodies that recognize specific carbohydrate epitopes in lipid extracts from different mouse tissues (Markotić et al. 1999). The importance of GSL phenotype of normal lymphoid cells has been well established in biochemical studies, and anti-GSL antibodies have become a major tool for their detection (Müthing 1996), but this was rarely coupled with histological and cellular analysis (Nakamura et al. 1995). The antibodies used in this study effectively localized biochemically determined GSLs in cryosections of mouse skeletal muscle (ČaČić et al. 1994), human skeletal and heart muscle (ČaČić et al. 1995), and human brain (Heffer–Lauc et al. 1996).
According to our data, mature thymocytes expressed a more restricted repertoire of GSLs compared to immature thymocytes. Immature T-lymphocytes bound all tested anti-GSL antibodies, except Gb4Cer antibody. Mature CD3+high thymocytes retained the expression of Gg4Cer and GalNAc-GM1b epitope and acquired the expression of Gb4Cer epitope.
During prenatal development, thymocytes change their ganglioside profile, shifting from a high expression of GM1a-type gangliosides towards GM1b-type gangliosides (Noguchi et al. 1994). GM1b-type gangliosides have been biochemically characterized as major gangliosides in the mature T-cells. In the biosynthesis of GM1b-type gangliosides (“asialo-pathway”) LacCer is elongated by a specific GalNAc-transferase to form Gg3Cer (Figure 1; Iber et al. 1992; van Echten and Sandhoff 1993). Further elongation to Gg4Cer is carried out by galactosyl-(β1–3)-transferase, and sialyGg4Cer by sialyltransferase-IV leads to the formation of GM1b, a ganglioside characteristic of mouse spleen (Figure 1; Nakamura et al. 1987). GM1b is particularly expressed by mouse splenic T-lymphocytes (Figure 1; Müthing et al. 1987) and thymocytes (Figure 1; Müthing et al. 1989). We were not able to detect GM1b ganglioside, because in thin-layer chromatography GM1b is recognized by anti-Gg4Cer antibody after Vibrio cholerae neuraminidase treatment (Müthing 1996). The Gg4Cer backbone of neutral GSLs present in the tissue would therefore interfere with ganglioside detection in histological sections or cell preparations. Further elongation of GM1b by the addition of β-linked N-acetylgalactosamine to position 4 of the terminal galactose appears to be coupled with T-cell activation and leads to the formation of GalNAc-GM1b, a T-cell differentiation-associated ganglioside (Figure 1; Horikawa et al. 1991; Müthing et al. 1989; Müthing 1997). Immunohistochemical and flow cytometric detection of GalNAc-GM1b on immature thymocytes in our study is not in accordance with the previous biochemical studies that assigned GalNAc-GM1b specifically to mature and activated T-lymphocytes (Figure 1; Müthing 1997). This can be explained, at least in part, by antibody crossreactivity to protein antigens, as confirmed by Western blotting analysis of protein extracts from the thymus.
The anti-Gb4Cer antibody was the only one that specifically bound to mature thymocytes, in accordance with biochemical studies that characterized globoside as a marker for small subpopulation of mature T-cells (Figure 1; Mühlradt et al. 1984). The most intense anti-Gb4Cer antibody staining was at the corticomedullar junction, where negative thymocyte selection is believed to be most stringent and where high endothelial venules (HEV) are located (Janeway and Travers 1996). The onset of Gb4Cer epitope expression, which starts after the thymocytes have completed all selection events, may therefore be involved in the export of mature cells through HEV.
T-lymphocytes from the secondary lymphoid organs bound only anti-GalNAc-GM1b and anti-Gg4Cer antibodies, whereas other GSL structures were detected only on a minor proportion of cells, confirming that T-lymphocytes maintain their GSL phenotype after maturation and release from the thymus (Figure 1). However, there was a difference in the distribution and level of expression of GalNAc-GM1b epitope between the spleen and the lymph nodes. Anti-GalNAc-GM1b antibody strongly stained T-cell-dependent periarteriolar sheets in the spleen, and homogeneously stained the entire CD3+ population in flow cytometry. In contrast, staining of the lymph node with this antibody was less intense and diffuse. Flow cytometry detected two populations of GalNAc-GM1b-positive cells: a smaller one with high fluorescence intensity staining (indicating high density of the epitope on the cell surface), comprising 5–10% of lymph node lymphocytes, and a larger one with low cellular fluorescence. It is difficult to explain these differences, especially in view of the finding that anti-GalNAc-GM1b antibody also recognized a specific glycoprotein in a Western blot. It is possible that the functional differences between the cellular microenvironments of these two lymphoid organs contribute to the observed differences. The spleen and the lymph nodes differ in the way of presenting an antigen, entering the lymph node via the lymph and the spleen from the blood (Janeway and Travers 1996). Consequently, the spleen and the lymph node differ in the type and distribution of stromal cells, including antigen-presenting cells, which may influence the GSL profile of the organ microenvironment.
The entire B-lymphocyte population from secondary lymphoid organs was recognized by all anti-GSL antibodies tested. This indicates that mature, unstimulated murine B-lymphocytes do not have a specific GSL profile as do T-lymphocytes. The possibility that such a broad positivity could be due to the polyclonal character of the antibodies, i.e., nonspecific recognition of surface immunoglobulins, has been ruled out by complete absence of binding of control preimmune sera in the dilutions used for immunohistology or flow cytometry. Moreover, the polyclonal anti-Gb3Cer antibody used in this study has the same specificity as anti-Gb3Cer monoclonal antibodies on human tonsil lymphocytes (Mangeney et al. 1991; Miyamoto et al. 1997), labeling exclusively a B-cell population comprising about 12–14% of human tonsil lymphocytes (our unpublished results). These results point to the interspecies differences in GSL expression between murine and human lymphocytes. Human B-lymphocytes express a more restricted GSL repertoire, so that specific subpopulations can be detected using antibodies directed to different neutral GSLs (Madassery et al. 1991; Wiels et al. 1991; Miyamoto et al. 1997), whereas murine B-lymphocytes have less specific GSL profiles.
Anti-GSL antibodies stained B-cell-dependent zones of the secondary lymphoid organs even after lipid extraction with chloroform/methanol, suggesting that non-lipid epitopes were recognized by these antibodies. However, Western blotting analysis revealed a specific acceptor glycoprotein only for the GalNAc-GM1b sequence. Binding of anti-GalNAc-GM1b antibody to a specific protein is a novel finding and requires further analysis concerning the biological significance and biochemical character of the detected protein. This protein may be responsible for immunoreactivity of immature thymocytes and B-lymphocytes which, according to previous biochemical studies, were not expected to react with GalNAc-GM1b antibody. GalNAc-GM1b antibody did not detect the GalNAc-GM1b sequence in a biochemical analysis of ganglioside fractions from cultured B-cells (Pörtner et al. 1993), indicating that strong binding of GalNAc-GM1b antibody to B-lymphocytes could indeed be due to a carbohydrate sequence on a specific glycoprotein. Although antibodies raised to GSLs have been considered to rarely cross-react with glycoproteins (Schlosshauer et al. 1988; Yang et al. 1994), several studies showed that carbohydrate chains linked to glycoproteins and GSLs may be responsible for simultaneous recognition by anti-GSL antibodies. For example, GM3 oligosaccharide Neu5Acα3Galβ4Glc-R is structurally almost homologous with the oligosaccharide Neu5Acα3Galβ4GlcNAc-R, so that anti-GM3 antibody crossreacts with glycoproteins (Müthing et al. 1994c). In addition, Galα1–4Gal disaccharide, responsible for the recognition of the CD77 molecule (Gb3Cer), could be simultaneously recognized as a glycoprotein-bound determinant (Kirkeby et al. 1998).
Despite the fact that there was no specific staining of protein extracts by other anti-GSL antibodies in the Western blotting analysis, most antibodies stained tissue cryosections even after lipid extraction with chloroform and methanol, which should remove all lipid-specific binding. These discrepancies can be explained by differences in tissue preparation for Western and immunohistochemical analysis. GSLs may be insoluble in detergents or organic solvents because of their membrane compartmentalization (Hakomori et al. 1998), which would make them resistant to chloroform/methanol extraction in cryosections. Another possible explanation for the negative Western blots with anti-GLS antibodies may lie in the loss of proteins during a standard Western blotting procedure (Gallagher and Smith 1993), especially proteoglycans or glycosaminoglycans of the extracellular matrix, which may carry crossreactive structures but require special solubilizing techniques during protein extraction (Esko 1993).
For some antibodies, there was a discordance in immunohistochemical and flow cytometric data. For example, GalNAc-GM1b was detected on almost all thymocytes by flow cytometry, whereas in immunohistochemistry it was almost undetectable in the thymic cortex, which harbors maturing T-lymphocytes. In addition, anti-Gg4Cer and anti-Gb4Cer antibodies did not react with B-cell-dependent zones in the secondary lymphoid organs, whereas they bound to B-cells in flow cytometry. A possible explanations for such discrepancy is a change in the surface density and composition of membrane GSLs and gangliosides during preparation of single-cell suspensions, which may affect recognition by anti-GSL antibodies (Nores et al. 1987; Rösner et al. 1990; Lloyd et al. 1992; Hildebrandt 1996; Tatewaki et al. 1997). Another explanation could lie in the influence of the fixative on the reliability of immunohistochemical studies (Prasadarao et al. 1990; Schwarz and Futerman 1997). Moreover, GSLs are located not only in the plasma membrane but also at subcellular levels (Gillard et al. 1993), and immunohistochemically stained sections reveal intracellular as well as surface antigens, whereas flow cytometry performed on viable, non-permeabilized cells gives information only about surface expression of antigens. Cells within the tissue are embedded in specific stromal elements that could also harbor carbohydrate antigens or, alternatively, mask carbohydrates expressed on the cell surface. Such elements could be lost during the preparation of single-cell suspensions and could therefore either increase antibody binding by exposing the cell surface or decrease it by shedding off specific epitopes. Differences in GSL accessibility of single cells and tissue-embedded cells may also reflect the differences between circulating lymphocytes in the body and resident cells in secondary lymphoid organs. The consequence of this idea is that cells might change their GSL accessibility according to their biological function throughout their lifespan by yet unknown mechanisms.
Despite these methodological issues, which require experimental clarification, our study clearly showed that murine T- and B-lymphocytes differ in their expression of glycolipids and that immunohistochemistry and flow cytometry using biochemically well-characterized GSL antibodies may provide important information complementing standard biochemical analyses of glycolipids.
Footnotes
Acknowledgements
Supported by a research grant from the Croatian Ministry of Science and Technology (“Inflammation in the nervous system, the role of cytokines and chemokines,” no. 1080110, A. Marušić), a grant from the Deutsche Forschungsgemeinschaft (DFG, SFB 549 “Macromolecular Processing and Signaling in the Extracellular Matrix,” project B07, J. Müthing), and was performed under the framework of a bilateral scientific cooperation between Germany and Croatia (BMBF project KRO-002-99).
We also thank Dr D. Batinić (Zagreb University School of Medicine) for critical help during this study and Dr R. Antolović (Pliva Research Institute) for critical help with the Western blotting analysis, Ms š. Čavar for excellent technical assistance with flow cytometry, and Ms Baranski and Dr M. Krohn (International Bureau of the BMBF) for administrative help.