
Abstract
Fluorescent immunocytochemistry (FICC) allows multiple labeling approaches when enzyme-based techniques are difficult to combine, such as in double-labeling experiments targeting small-caliber axonal segments. Nevertheless, the conversion of FICC to a product visible at the electron microscopic (EM) level requires labor-intensive procedures, thus Justifying the development of more user-friendly conversion methods. This study was initiated to simplify the conversion of FICC to EM by employing the unique properties of tyramide signal amplification (TSA), which allowed the simultaneous targeting of a fluorescent tag and biotin label to the same antigenic site. Briefly, one of two antigenic sites typically co-localized in damaged axonal segments was visualized by the application of a fluorescent secondary antibody, with the other tagged via a biotinylated antibody. Next, an ABC kit was used, followed by the simultaneous application of fluorophore-tyramide and biotin-tyramide. After temporary mounting for fluorescent digital photomicroscopy, sections were incubated in ABC and reacted with diaminobenzidine before EM analysis. Double-labeling fluorescent immunocytochemistry with TSA clearly delineated damaged axonal segments. In addition, these same axonal segments yielded high-quality EM images with discrete electron-dense reaction products, thereby providing a simple and reproducible means for following fluorescent analysis with EM.
T
Fluorescent markers are more appropriate for such studies. However, their use necessitates further conversion methods when subsequent electron microscopic (EM) investigations are needed to confirm their precise sites of localization (Sandell and Masland 1988; Lubke 1993; Todd 1997; Humbel et al. 1998). Recently, several conversion methods have been described, including photoconversion of fluorescent tracers with DAB (Sandell and Masland 1988; Lubke 1993), the use of anti-fluorescein antibodies combined with streptavidin-gold (Shiosaka et al. 1986) or immunogold (Luby-Phelps et al. 1984), and the use of fluorescent tags directly (Powell et al. 1997) or indirectly (Powell et al. 1998) coupled with gold particles. Although the use of laser scanning confocal microscopy (LSCM) has offered new possibilities for the conversion of fluorescent multiple labeling ICC to EM (Sun et al. 1995; Todd 1997), the technique is rather complex and the more restricted availability of the instrument limits its employment. Therefore, the vast majority of present techniques that allow combined fluorescent/ultrastructural visualization require time-consuming and labor-intensive procedures and/or the use of special technical tools that constitute obstacles to their routine laboratory use.
Recently, in our laboratory we have successfully utilized a combination of the peroxidase-based chromogens benzidine dihydrochloride (BDHC) and diaminobenzidine (DAB) or Vector VIP for both LM and EM co-localization of reaction products linked to calcium-induced, calpain-mediated spectrin proteolysis (CMSP) and cytoskeletal change in traumatically injured axonal segments (Büki et al. 1999a). However, despite our success in these approaches, we wished to utilize double-label fluorescent approaches that would provide better chromogen penetration and higher spatial resolution and reliability while offering a more simplified method of processing and evaluation than the use of enzyme-based chromogenic immunohistochemical techniques. Having demonstrated that traumatic axonal injury leads to activation of the neutral protease calpain, while also establishing the spatiotemporal correlation between local intra-axonal CMSP and neurofilament compaction (NFC) as markers of traumatic axonal injury (Povlishock et al. 1997; Büki et al. 1999a,b; Okonkwo et al. 1999), we sought to utilize these specific changes as targets for new LM double labeling, a fluorescent approach that is also compatible with EM analysis. Our goal was to improve the sensitivity and utility of our experimental methods, thereby saving time and, more importantly, reducing the number of animals required for unraveling the pathobiology and treatment of traumatic axonal injury.
Recognizing that the TSA technique allows the simultaneous application of a fluorescent tag [fluorophore (rhodamine)-tyramide] and biotin (biotin-tyramide) to the same antigenic site, we describe here a substantial modification of the conventional TSA technique that yields a reliable and simple method that can be employed for fluorescent double/multiple-labeling experiments combined with subsequent pre-embedding immunoelectron microscopic analysis of the labeled structures. We also comment on the utility of this method in the study of axonal profiles, in which it combines the advantages of sensitive signal amplification with a user-friendly conversion method for immunoelectron microscopy.
Materials and Methods
Induction of Traumatic Brain Injury
For induction of focal axonal injury, a well-characterized rodent model of impact acceleration head injury was employed as described by Marmarou et al. (1994). Eight Sprague-Dawley rats (Charles River Laboratories; Raleigh, NC) weighing 368-396 g were used for the experiments. For induction of anesthesia, the animals were exposed to 4% isoflurane (Iso Flo; Abbott Laboratories, North Chicago, IL) in a bell jar for 5 min and then intubated and ventilated with a mixture of 1-2% isoflurane in 30% O2 and 70% N2O. Next, the skull between the coronal and lambdoid sutures was exposed with a midline incision. A metallic disk-shaped helmet 10 mm in diameter was firmly attached to this point of the skull using dental acrylic. The animal was placed in a prone position on a foam bed with the metallic helmet centered under the edge of a plexiglas tube and was prevented from falling by two belts secured to the foam pad. Brass weights weighing 450 g were allowed to fall from a height of 200 cm through the plexiglas tube directly to the metallic disk fixed to the animal's skull, a setting that produces neither cerebral contusion nor subdural hemorrhage. After the injury the animal was immediately ventilated with 100% O2. The helmet was removed and the skull was investigated for any sign of fracture which, if found, would have disqualified the animal from further evaluation. The scalp wound was sutured, with the animal remaining on artificial ventilation until it regained spontaneous breathing, followed by sacrifice at the predetermined survival periods of 30 (three rats) and 180 min (three rats) after injury. Two sham-injured animals (one for each survival period) were treated in the same way as the other three animals per group but were not injured.
Physiological Assessments
The respiratory status was monitored through pulse oximetry via the footpad and/or the tongue. In addition, brain temperature was monitored by a temporalis muscle probe and core temperature was determined by a rectal probe.
Immunohistochemistry
At the designated survival time, the rats were reanesthetized with an overdose of sodium pentobarbital and transcardially perfused with 4% paraformaldehyde and 0.1% glutaraldehyde in Millonig's buffer. The brains were immersed in the same fixative overnight (16-18 hr). On the basis of previously published observations concerning the topography of diffusely injured axons in rat brain (Povlishock et al. 1997), a midline 5-mm-wide block of the brain containing the medullary pyramids was removed using a sagittal brain blocking device to include the region extending from the interpeduncular fossa to the first cervical segment. The tissue was sectioned with a Vibratome Series 1000 (Polysciences; Warrington, PA) at a thickness of 30 μm and was collected in 0.1 M phosphate buffer. Then the sections were rinsed three times for 10 min in PBS, placed in an Na-citrate buffer (pH 6.0), and transferred to a programmable magnetron-powered 900-W PELCO 3460 microwave oven (Ted Pella; Redding, CA). This laboratory microwave oven is equipped with a load cooler and a computerized temperature monitoring system, and was operated with 70% energy cycling over two 5-min periods during which tissue temperature was never allowed to rise above 40C. This approach was based on our recent finding (Stone et al. 1999) that the use of microwave energy without the generation of considerable heat significantly enhances immunoreactivity while also suppressing background staining. This controlled temperature approach also allows the preservation of excellent ultrastructural immunohistochemical detail, unlike traditional approaches that may cause tissue damage via the generation of significant heat (Stone et al. 1999). After microwave processing, sections were incubated for 35 min with 0.2% Triton X-100 (Sigma Chemical; St Louis, MO) in a 10% mixture of normal goat serum (NGS) and normal donkey serum (NDS) (Sigma) in PBS. After two quick rinses in PBS containing 1% NGS/NDS, sections were incubated with a mixture of the primary antibodies, Ab38 and RMO-14, overnight (12-16 hr). The rabbit polyclonal antibody Ab38 (applied here at a dilution of 1:18,000) targets the NH2-terminal fragment of the α-subunit of brain spectrin on its cleavage by the calcium-activated protease calpain, allowing the identification of the precise locus of any calcium-mediated events linked to the proteolysis of spectrin at both the LM and EM level (Siman et al. 1989; Roberts-Lewis et al. 1994). The mouse monoclonal antibody RMO-14 (1:500) has been described to exclusively target rod domains of compressed neurofilament M-subunits (NF-M) visualized only on alteration of their side arms, with subsequent exposure of their previously masked epitopes (Lee et al. 1987; Pettus et al. 1994; Povlishock et al. 1997; Okonkwo et al. 1998). Because such NFC and side-arm modification routinely occurs in foci of traumatically induced axonal damage (Maxwell et al. 1997; Povlishock et al. 1997; Okonkwo et al. 1998), this antibody delineates the precise sites of axonal injury in which the activated CMSP is also found (Büki et al. 1999a).
After rinses in 1% NDS/NGS, sections were incubated in a solution of coumarin-labeled donkey anti-mouse immunoglobulin (1:200; Jackson Immunoresearch Laboratories, West Grove, PA) and biotinylated goat anti-rabbit immunoglobulin (1:400; Vector, Burlingame, CA) for 60 min, followed by three 10-min rinses in PBS. In all subsequent steps of tissue processing, direct illumination of the sections was avoided.
Tyramide Signal Amplification
Based on attempts to simplify the TSA method and make it more cost-effective, while also providing better preservation of ultrastructural detail several modifications of the original protocol (for details see the commercial protocol, Renaissance Kit, NEN Life Sciences Products, Boston, MA) were introduced in our laboratory. Specifically, after incubation in an avidin-biotin-peroxidase complex (ABC standard Elite kit; Vector; dilution 1:200) with rinsing in TNB blocking buffer (Renaissance Kit, NEN Life Science) for 20 min and in PBS twice for 10 min, a 1:1 mixture of rhodamine-tyramide and biotin-tyramide (Renaissance Kit, NEN Life Science), both diluted 1:300, was applied. A 1:6 mixture of amplification diluent (Renaissance Kit, NEN Life Science) and PBS constituted the solvent for the tyramide stock solution, together forming the working solution. The working solution was applied in mid-sized tissue culture wells containing six tissue sections each (12-well/tray; Falcon Multiwell Tissue Culture Plate, Becton Dickinson Labware; Becton Dickinson, Lincoln Park, NJ), 400 μl working solution/well, for 12 min, followed by three 10-min rinses in PBS. The dilution of the amplification diluent in PBS to 1:6, as well as the substitution of the detergent-containing rinsing buffer (Tween is suggested in the original protocol) with PBS was aimed to prevent unnecessary membrane damage and reduce the background reactivity. Next, the sections were mounted on premium microscope slides (Fisher Scientific; Pittsburgh, PA) and coverslipped using 50% glycerol dissolved in double-distilled water. The slides were immediately transferred to a Nikon Eclipse E 800 biological research microscope (Image Systems; Columbia, MD) equipped with a Sony Catseye digital camera (Image Systems). Images of immunofluorescent axonal profiles were digitally captured and archived. All the sections analyzed were mounted and coversliped separately, with each assigned a serial ID number. After completion of digital acquisition, coverslips were removed and the sections were re-rinsed and incubated in an avidin-biotin-peroxidase complex at a dilution of 1:200. The sections were then processed for visualization of the immunohistochemical complex using 0.05% diaminobenzidine (DAB) (Sigma) and 0.01% H2O2 in 0.1 M phosphate buffer. Although in theory one could argue that the previously applied peroxidase molecule found in the ABC complex, used to activate tyramide, could also serve as the catalyst for the chromogenic reaction with DAB, this approach consistently yielded such a weak signal as to preclude its usefulness for routine LM/EM studies.
Ultrastructural Analysis
After the above prepared sections were dehydrated and flat-embedded between plastic slides in Medcast resin (Ted Pella), areas for EM investigation were selected by repeated, detailed analysis involving comparisons with the previously captured digital images. To draw a precise correlation between these samples assessed either with fluorescence or routine light microscopy, we employed anatomic landmarks such as the vicinity of blood vessels, tissue edges, and the relative distance between labeled axonal segments. Once identified, homologous immunopositive foci prepared for ultrastructural analysis were trimmed, mounted on plastic studs, and sectioned using an LKB Ultratome at a thickness of 70-100 nm. A few semithin sections were mounted and cover-slipped for comparison to the digital images while also serving as topographic guides for the ultrastructural analysis. Thin sections were picked up on Formvar-coated slotted grids, then stained in 5% uranyl acetate in 50% methanol for 2 min and 0.5% lead citrate for 1 min. Alternatively, some sections were not stained to provide additional evidence for antibody specificity. Ultrastructural analysis was carried out using a JEOL-1200 electron microscope.
Immunohistochemical Controls
The antibodies employed have been extensively characterized in the existing literature and widely used (Lee et al. 1987; Roberts-Lewis et al. 1994; Saatman et al. 1996; Posmantur et al. 1997; Povlishock et al. 1997; Büki et al., 1999a). Nevertheless, additional control studies were performed to achieve further rigor. To this end, all immunocytochemical reactions included sections incubated without the addition of the primary or secondary antibody or with the secondary alone with or without TSA in concert with multiple dilution strategies to obtain optimal staining.
Results
Fluorescent and Routine LM Observations
In accord with the results of our previously published studies, traumatically injured axonal profiles displayed focal axonal immunofluorescence representing neurofilament compaction (RMO-14) and calpain-mediated spectrin proteolysis (Ab38), with the highest proportion of these profiles found in the corticospinal tract and in the medial longitudinal fasciculi (Povlishock et al. 1997; Okonkwo et al. 1998,1999; Büki et al. 1999a,b). Thirty min after injury the immunoreactive axonal segments appeared sharply delineated as swollen and occasionally vacuolated profiles. At 180 min after injury the majority of the immunolabeled axonal foci appeared as large, swollen axonal bulbs indicating axonal disconnection. The majority of the visualized axonal profiles displayed both co-localized coumarin immunofluorescence, representing NF alteration, and rhodamine fluorescence, delineating calpain-mediated spectrin proteolysis at all time points examined. In addition, the localization of the individual fluorescent markers was easier to recognize and more precise than that achieved by the previously employed peroxidase enzyme-based intra-axonal chromogens. Specifically, by applying a 2.5- to fourfold lower concentration of the primary antibody (Ab38) than employed in traditional immunohistochemical investigations and a sixfold lower concentration of the tyramide solution than originally recommended for this technique, we recognized rhodamine-TSA to be precisely localized to the focus of the axonal damage, without background immunoreactivity in any other tissue elements and/or emission spectra. Furthermore, this protocol did not compromise the sharp detection of isolated axonal profiles single labeled with the RMO-14 antibody (coumarin fluorescence) but not labeled with rhodamine, a finding also consistent with our previous observations (Büki et al. 1999a) (Figures 1A–1C). The digital comparison of homologous fluorescent and brightfield images revealed the consistent localization of the reaction products and also confirmed the accuracy of the conversion method itself. Specifically, the DAB reaction product (Figure 1D) was precisely localized to the same sites containing rhodamine labeling. Furthermore, none of the single-labeled axonal profiles displaying NFC alone were DAB-positive, emphasizing the reliability of the method chosen.
Ultrastructural Observations
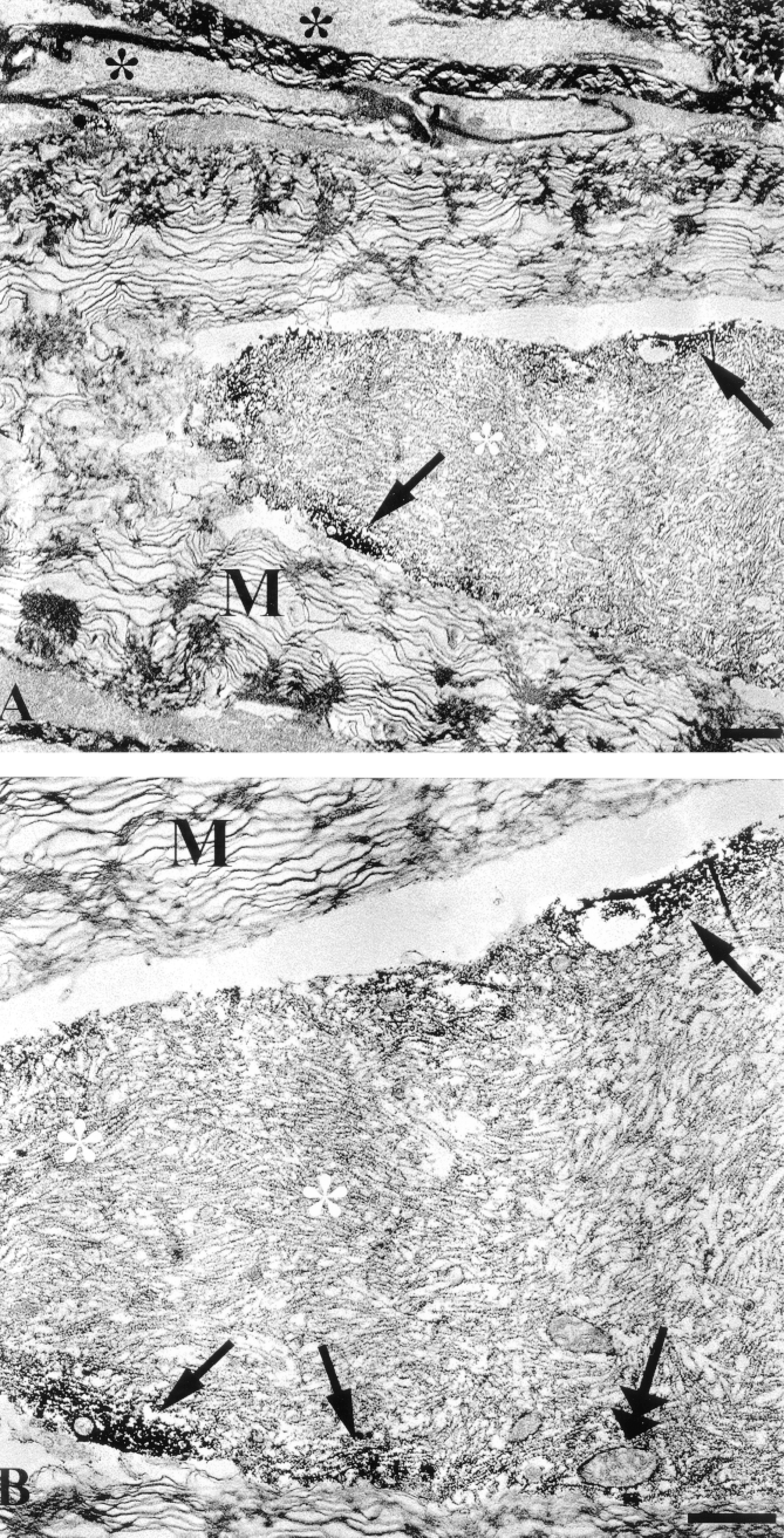
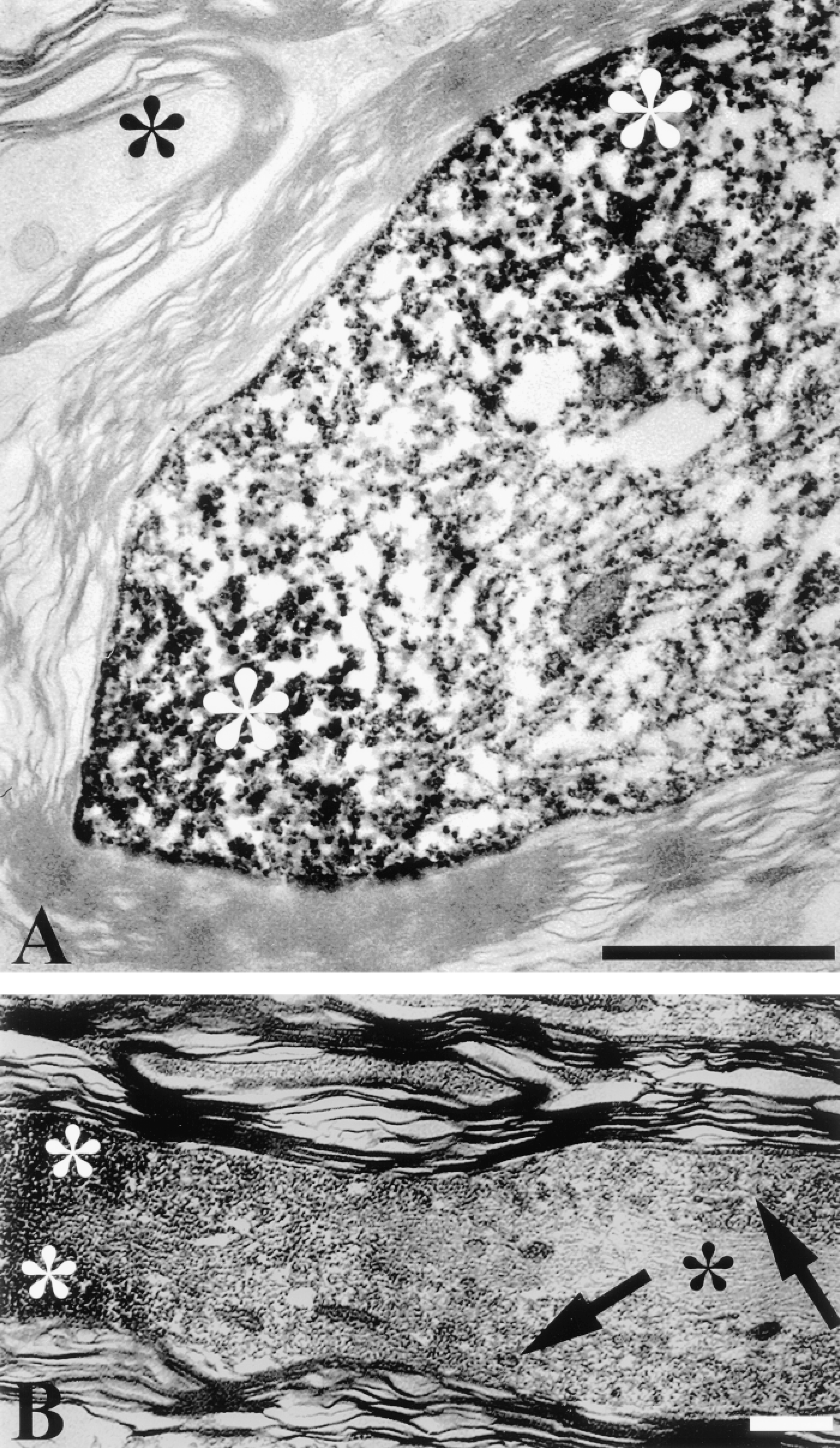
In this approach, the overall ultrastructural appearance of the tissue described above was well-preserved despite these additional tissue-processing procedures. At the EM level, damaged immunoreactive axons displayed the well-established repertoire of traumatic axonal injury, including neurofilament compaction, mitochondrial swelling, loosening of the myelin sheath, and formation of periaxolemmal spaces (Christman et al. 1994; Maxwell et al. 1997; Povlishock et al. 1997) (Figures 2A, 2B, 3A, and 3B). CMSP immunoreactivity was observed as an electron-dense DAB reaction product in axonal foci where neurofilaments were clearly compacted. At 30 min after injury the electrondense DAB granules were predominantly localized in the subaxolemmal compartment (Figures 2A and 2B), whereas at 180 min after injury the altered cytoskeletal elements in the entire damaged axonal segment were covered with reaction product (Figures 3A and 3B). The TSA method provided excellent sensitivity and specificity at the ultrastructural level, with the immunoreactivity exclusively localized in the injured axonal segments and with no spreading or diffusion into non-injured axons (Figures 2 and 3). Moveover, the ultrastructural appearance of the DAB reaction product was identical to or perhaps even more intense than that detected in our previous studies (Povlishock et al. 1997; Büki et al. 1999a), indicating that neither signal amplification or the slightly electron-dense tyramide itself complicated the detailed ultrastructural analysis of immunoreactive damaged axons.
Immunohistochemical Controls
The exclusion of either the primary or the secondary antibody from the immunohistochemical reaction resulted in lack of immunoreactivity. Gradual overdilution of the immunosera caused disappearance of the immunostaining. Simultaneous or sequential application of immunosera in the case of double labeling F-IHC resulted in equally powerful detection of both antigens. The TSA kit alone did not contribute to any specific immunohistochemical reaction that might have influenced the analysis. Elimination of the rhodaminetyramide step resulted in the lack of fluorescent labeling of CMSP-IR axonal profiles, while the absence of biotin-tyramide resulted in the lack of specific DAB staining (i.e., lack of conversion) at the LM level without affecting the fluorescence signal (rhodamine label) indicating CMSP-IR.
Discussion
In a series of labor-intensive studies employing the combination of enzyme-based immunohistochemical reaction products (DAB/VVIP and BDHC/VVIP), we have recently demonstrated the co-localization of calpain-mediated spectrin proteolysis (CMSP) with markers of traumatically induced cytoskeletal alteration, providing evidence for the contribution of CMSP to the pathogenesis of traumatically induced axonal injury (TAI) (Büki et al. 1999a). In this study we utilized these observations as a template for establishing a new double-labeling light microscopic technique that was also compatible with EM investigation of the doublelabeled axonal profiles. In this search for a double-labeling method to provide excellent spatial resolution at both light and electron microscopic levels, we considered the application of immunofluorescent markers. However, we realized that despite their excellent spatial resolution, the conversion of immunofluorescent signals to EM was inherently difficult. This limitation was overcome by our recognition of the advantages of the recently developed TSA technique, also described as catalyzed reporter deposition (Bobrow et al. 1989,1992; de Haas et al. 1996; Hunyady et al. 1996; van Gijlswijk et al. 1996,1997; Van Heusden et al. 1997). Essentially, in the TSA technique the biotin-or fluorophore-conjugated tyramide is “attracted” to the antigenic site activated by the peroxidase component of the immunolabeling complex. The activated tyramide instantly and covalently binds to nearby tyrosine molecules, and this single enzymatic reaction greatly (1-400-fold) amplifies the signal intensity of the immunocytochemistry (Bobrow et al. 1989,1992; Hunyady et al. 1996; Erber et al. 1997; van Gijlswijk et al. 1997). Soon after the technique was first described, its widespread utilization (Van Heusden et al. 1997; Kaufmann et al. 1998; Kressel 1998; van de Corput et al. 1998; Speel et al. 1998,1999) and further modifications in its application highlighted its usefulness and importance, particularly in paradigms involving fluorescent double labeling with antibodies derived from the same species (Hunyady et al. 1996; Shindler and Roth 1996; Teramoto et al. 1998). Recently, this technique has also been used for pre- and postembedding immunoelectron microscopy, an extremely labor-intensive approach (Schofer et al. 1997; Stanarius et al. 1997; Punnonen et al. 1999), with some suggesting that the electron-dense tyramide can also be used for EM without concomitant DAB chromogen application (Mayer and Bendayan 1997).
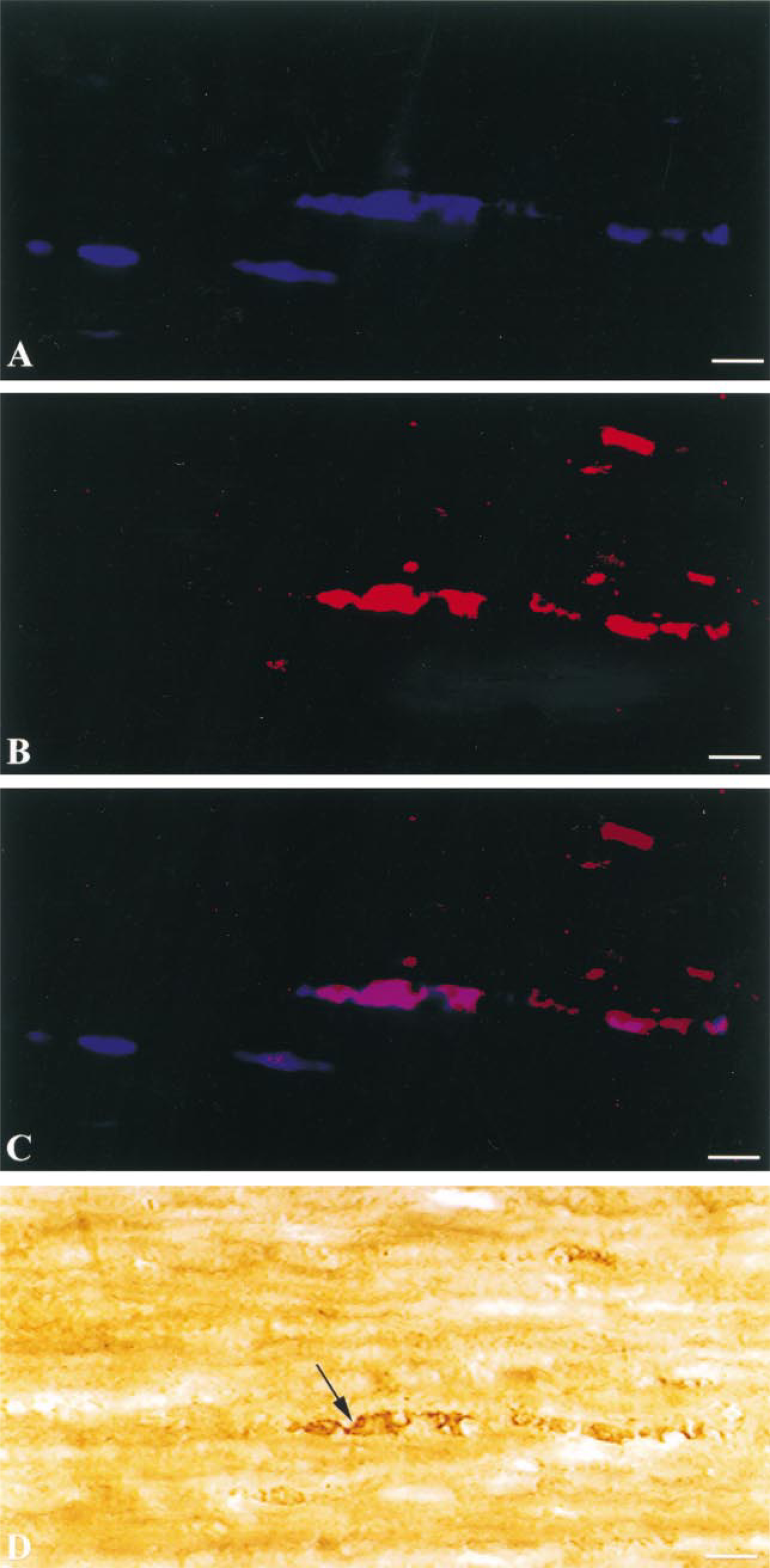
Fluorescent light micrographs demonstrating coumarin-labeled (
(
Tyramide signal amplification of the immunohistochemical reaction reliably demonstrates calpain-mediated spectrin proteolysis in axonal foci undergoing diffuse axonal injury 3 hr after injury (
In the present study, the combination of the conventional immunofluorescence (coumarin-labeled marker for neurofilament compaction) and fluorophore-labeled tyramide (rhodamine marker for calpain-mediated spectrin proteolysis) yielded high-quality fluorescent labeling that consistently denoted sites of injury previously described in this model of TAI. Further, the conversion of the tyramide label to an electron-dense DAB reaction product allowed this same fluorescently tagged structure to be easily carried forward to the EM level. We emphasize that the reaction presented here does not involve a direct conversion of the rhodamine tag but rather proceeds in relation to the co-localized biotin-tyramide. This strategy provided extremely high resolution and fidelity because virtually all the axonal profiles detected with the rhodamine tag were also labeled with DAB. It should be noted that none of the axonal profiles displaying single labeling with the coumarin marker alone demonstrated DAB reactivity. This finding further underlines the specificity and fidelity of the technique. As noted, at the ultrastructural level the tissue was well preserved, with the suggestion that the intensity of the staining was even more pronounced than that achieved through the tradititional HRP-DAB methods. Although we are uncertain as to why this was the case, we postulate that the concomitant presence of the electron-dense tyramide molecule itself enhanced the density of the overall reaction product (Mayer and Bendayan 1997).
In relation to the observed electron-dense reaction, we also note that the reaction product appeared to be of consistent size and density, showing consistent localization. To be candid, we were surprised by this finding because the histochemistry underlying tyramide amplification involves a tyramide-tyrosine covalent binding that is not directly determined by immunological principles (Bobrow et al. 1989,1992; van Gijlswijk et al. 1996,1997). In this scenario, one would predict a more widespread electron-dense reaction product, reflecting more random amplification.
Why our method is not confounded by this liability is unknown. However, we speculate that our modification of the original commercially available TSA protocol at several key steps may have provided optimal signal-to-noise ratio at both the LM and EM level while achieving an optimal cost-benefit ratio.
Also of note in this process is the fact that the simultaneous visualization of the biotin molecule by the DAB chromogen enables the investigator to overcome the problem of fluorescent signal fading over time.
We believe that our study convincingly demonstrates the versatility of the TSA method. Therefore, in addition to being an excellent technique for augmenting immunocytochemical reactions, it is also considered an outstanding tool for immunofluorescent double/multiple labeling, with the possibility of a simple signal conversion method for EM. Furthermore, because the simultaneous application of biotin- and rhodamine-tyramide facilitates ultrastructural analysis of double-labeled immunofluorescent profiles, this opens up new potential for the future use of this sensitive, versatile signal amplification technique.
Footnotes
Acknowledgements
Supported by grants NS 20193 and by the Martin Rod-bell Fellowship.
We thank Dr Robert Siman for kindly donating the Ab38 and Dr John Q. Trojanowski for the RMO-14 antibody. We also thank Lynn Davis, Thomas Coburn, and Judy Williamson for excellent technical support.