
Abstract
To elucidate the role of intermediate filament proteins in endocrine cells, we investigated the expression and subcellular distribution of GFAP in mouse islets of Langerhans. For this purpose, combined immunocytochemical and biochemical analysis with a panel of antibodies was carried out to identify GFAP-immunoreactive cells in mouse endocrine pancreas. Cell fractionation into NP-40-soluble and detergent/high salt-insoluble components was performed to assess whether GFAP was located in the cytosolic and/or cytoskeletal compartments of immunoreactive cells. Immunoelectron microscopic analysis was carried out to determine the subcellular distribution of the protein. Peripheral islet cells were stained with anti-GFAP antiserum. These cells were identified as glucagon-secreting cells by immunocytochemical staining of consecutive sections with anti-somatostatin, anti-GFAP, and anti-glucagon antisera. Western blotting analysis of both NP-40-soluble and detergent/high-salt insoluble fractions of isolated islets of Langerhans allowed detection of GFAP in both cytosolic and cytoskeletal compartments. Interestingly, however, the former location was highly predominant. In addition, immunoelectron microscopy localized GFAP associated with the periphery of secretory granules. On the basis of these results, an intriguing role for GFAP in secretory events should be strongly suspected.
T
The first step for IF polymerization is the formation of a dimer by the hydrophobic side-to-side interaction of two monomers whose rod domains intertwine in a coiled-coil fashion (Albers and Fuchs 1992; Fuchs and Weber 1994). Whereas CKs are obligate heterodimers, composed of Type I and Type II subunits (Albers and Fuchs 1992; Herrmann and Aebi 2000), vimentin-like (Type III) and lens (Type VI) IFs are usually homodimers (Steinert et al. 1993), even though vimentin has also been reported to heteropolymerize either with desmin or GFAP (Quinlan and Franke 1982, 1983). There is evidence of a co-assembling incompatibility between certain classes of IFs because of different axial dimensions and alignments (Steinert et al. 1993). Thus, for example, when a cell type displays either Type I/II or Type III IFs, these are arranged to form two distinct IF networks, thereby segregating CKs from Type III IFs (Osborn et al. 1980; Yoon et al. 1998; Herrmann and Aebi 2000).
For a long time, IFs have been considered very stable structures. This view, supported because of their resistance to solubilization by high-salt buffers and non-denaturating detergents (Quinlan and Franke 1983), has changed in the past decade in favor of a more dynamic model (Steinert and Liem 1990; Steinert et al. 1993; Herrmann and Aebi 2000). In addition to the polymerized form, a soluble pool of IF proteins (IFps) has been shown (Blikstad and Lazarides 1983; McTavish et al. 1983; Moon and Lazarides 1983; Soellner et al. 1985; Isaacs et al. 1989), and a growing body of evidence demonstrates that a dynamic steady-state exists between this “barely detectable” soluble pool and the much more represented polymerized IFps (Ip and Fellows 1990; Vickstrom et al. 1992; Miller et al. 1993; Okabe et al. 1993). Additional studies with green fluorescent protein-tagged cytoskeletal proteins and fluorescence recovery after photobleaching analyses clearly confirmed that IF networks are highly dynamic structures (Prahlad et al. 1998; Yoon et al. 1998). A major regulatory mechanism of the exchange between the soluble and polymerized forms of IFps appears to be protein phosphorylation (Steinert and Liem 1990; Fuchs and Weber 1994; Herrmann and Aebi 2000). In general, the more elevated the level of IF phosphorylation, the more the soluble pool of IFs increases (Chou et al. 1997; Liao and Omary 1996).
Cytoplasmic IFs, in general, show a developmentally regulated cell type-specific pattern of expression (Albers and Fuchs 1992; Steinert et al. 1993; Fuchs and Weber 1994; Herrmann and Aebi 2000). It is generally assumed, for example, that vimentin is expressed in vivo in cells of mesenchymal origin (Albers and Fuchs 1992; Fuchs and Weber 1994; Herrmann and Aebi 2000), desmin in muscle cells (Albers and Fuchs 1992; Fuchs and Weber 1994; Houseweart and Cleveland 1998; Herrmann and Aebi 2000), neuronal IFs in neurons (Albers and Fuchs 1992; Houseweart and Cleveland 1998; Herrmann and Aebi 2000), and CKs in epithelial cells (Albers and Fuchs 1992; Houseweart and Cleveland 1998; Herrmann and Aebi 2000). In this respect, GFAP, originally described in mature astrocytes (Eng et al. 1971), is considered a marker for certain glial cell types of the central and peripheral nervous systems (Yen and Fields 1981; Jessen et al. 1984; Fields and McMenamin 1985; Achtstätter et al. 1986a; Fuchs and Weber 1994). However, there is accumulating evidence that this pattern of expression is not as rigorous as it would seem. GFAP does not appear to be restricted to glial cells, and its expression has also been reported in a limited number of cell types of presumed mesenchymal origin scattered throughout several organs. Rat liver perisinusoidal stellate cells (Gard et al. 1985), rat Leydig cells (Holash et al. 1993), chondrocytes from human elastic cartilage (Kepes and Perentes 1988), and rat pancreatic periacinar stellate cells (Apte et al. 1998) are some examples. In a previous article, we reported that GFAP-like immunoreactivity could also be detected in epithelial cells, i.e., glucagon-secreting cells, of the rat pancreas (Regoli et al. 2000). In the present study, in addition to demonstrating the expression of GFAP in glucagon-secreting cells of mouse pancreas, we detected its subcellular distribution. In contrast to current views, biochemical cell fractionation procedures demonstrate that GFAP is mostly present in its soluble form and immunoelectron microscopic analysis of GFAP localization in glucagon-secreting cells shows that GFAP is associated with secretory granules. These findings therefore provide intriguing evidence that, under physiological circumstances, the ratio between soluble and polymerized forms of IFps is reversed in favor of the soluble pool, and that a direct involvement of IFps in the secretory mechanisms of glucagon granules can be envisioned.
Materials and Methods
Materials
Ficoll 400, Hank's balanced salt solution (HBSS), soybean trypsin inhibitor (STI), diphenilthiocarbazone, 3–3′ diaminobenzidine (DAB)-HCl, and poly-
Antibodies
A rabbit polyclonal antibody (PAb), anti-GFAP, raised using bovine spinal cord as immunogen, was purchased from Zymed Laboratories (South San Francisco, CA). A monoclonal antibody (MAb), anti-GFAP (clone 6F-2), was a kind gift from Dr. R. Salvestroni and Dr. S. Pluchino (Institute of Neurology, University of Siena, Siena, Italy). A rabbit PAb, anti-GFAP, raised using human brain as immunogen, was from Sigma and was generously offered by Dr. C. Nicoletti (Laboratory of Molecular Recognition, The Babraham Institute, Cambridge, UK). Alkaline phosphatase (AP)-conjugated anti-rabbit and anti-mouse IgG were obtained from Roche Diagnostics and Sigma-Aldrich, respectively. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG, HRP-conjugated goat anti-mouse IgG, sheep anti-rabbit IgG, and peroxidase–anti-peroxidase complex were from Sigma–Aldrich. Rabbit anti-glucagon and anti-somatostatin PAbs were a generous gift from Dr. M. Bendayan (Department of Pathology and Cell Biology, University of Montreal, Montreal, PQ, Canada). Twenty-nm gold-conjugated anti-rabbit IgG was from BioCell and was kindly provided by Dr. N. Volpi (Department of Biomedical Science, University of Siena).
Animals
Fifteen 1-year-old male C57 mice were used for this study. After ether anesthesia, animals were decapitated and the splenic portion of the pancreas, the intestines, and the brains were taken. Five pancreata, intestines, and brains were processed for light (LM) and transmission electron microscopy (TEM). The remaining 10 pancreata and brains were used for biochemical evaluations.
Islet Isolation Procedure
After enzymatic digestion of the pancreas, islets of Langerhans were purified using a discontinuous density gradient of Ficoll solutions as previously reported (Sutton et al. 1986), with minor adjustments. Briefly, the common bile duct was cannulated and 1 ml of digestion solution (1 mg/ml collagenase in HBSS containing 10 mmol HEPES) was injected to distend the pancreas. The pancreas was removed and placed for 15 min at 37C in a Petri dish filled with 5 ml of digestion solution. Then the pancreata were further minced with scissors and the pancreatic tissue was gently syringed through a 16-gauge needle. The digestion of the pancreas was further prolonged for 15 min with gentle shaking, and then the pancreatic tissue was poured into a 50-ml centrifuge tube, washed twice with 20 ml ice-cold washing medium (HBSS with addition of 10 mmol HEPES and 0.2 mg/ml STI), filtered through a double layer of gauze, centrifuged, and re-suspended in 25% Ficoll made up in washing medium (WM). This suspension was overlayered with a discontinuos density gradient of 23%, 20%, and 11% Ficoll solutions and centrifuged at 800 × g for 10 min in a swinging-bucket rotor at 4C. The pancreatic tissue present in the upper two interfaces was recovered, washed twice with WM, and re-suspended in 3 ml of the same medium with the addition of Dithizone (150 μl, 0.25 mM diphenylthiocarbazone in 1% DMSO/MEM/1% BSA) as previously reported (London et al. 1990). Dithizone-stained (red-colored) islets were picked up by hand under a dissection microscope and resuspended in 1 ml lysis buffer (10 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1 mM EDTA, 0.5% Nonidet NP-40, 2 mM PMSF, 50 μg/ml aprotinin) or in 4% paraformaldehyde dissolved in 0.1 M cacodylate buffer (pH 7.4).
Preparation of IF-enriched Cytoskeletons and Cell Fractionation Procedures
Tissues were fractionated into pelletable (insoluble) components and nonpelletable (soluble) proteins as previously reported (Achtstätter et al. 1986a), with minor modifications. Briefly, purified islets in lysis buffer were sonicated and the lysate was centrifuged at 10,000 × g for 20 min at 4C. The supernatant (NP-40-soluble fraction) was frozen and stored at −20C. The pellet was resuspended in high-salt buffer (10 mM Tris-HCl, 140 mM NaCl, 1.5 M KCl, 5 mM EDTA, 0.5% Triton X-100, pH 7.6, 2 mM PMSF, 50 μg/ml aprotinin, 1 mM 2-mercaptoethanol) and incubated for 1 hr at 4C with moderate spin magnetic stirring. At the end of this incubation, the suspension was centrifuged at 10,000 × g for 20 min at 4C.
The supernatant was discarded and the pellet was resuspended in the same volume of high-salt buffer and further incubated for 30 min at 4C with magnetic stirring. The pellet of cytoskeletal material obtained after centrifugation at 10,000 × g for 20 min at 4C was washed twice in 0.01 M PBS, pH 7.4, containing 1 mM 2-mercaptoethanol and was dissolved in 40 μl Laemmli's sample buffer for 6 hr at RT with gentle magnetic stirring. Finally, the sample was centrifuged at 20,000 × g for 30 min at 25C and the supernatant (IF-enriched fraction) was used for immunoblotting analysis.
Small cubes of brain tissue were homogenized in lysis buffer with a Potter glass–teflon homogenizer and treated with the above-described procedure.
Western Blotting
IF-enriched fractions and a 1:8 dilution of NP40-soluble fractions of islets and brain tissue were separated by electrophoresis on a 12% polyacrylamide gel according to Laemmli (1970), and transferred to nitrocellulose in a Bio-Rad Trans-blot apparatus as reported by Burnette (1981), using 25 mM Tris, pH 8.3, 192 mM glycine, 20% (v/v) methanol as blotting buffer. After washing nitrocellulose membranes with 10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% (v/v) Tween-20 (TBS-Tween), endogenous peroxidases were blocked with 3% (v/v) H2O2 in distilled water. Bovine serum albumin (BSA) (3%, w/v) in TBS–Tween (blocking buffer) was used to quench nonspecific protein adherence of the Abs. Membranes were incubated overnight in a 1:500 dilution of rabbit anti-GFAP PAb in blocking buffer or in a 1:200 dilution of anti-GFAP MAb in blocking buffer. GFAP in IF-enriched cytoskeletal fractions was detected by incubating membranes with the appropriate HRP-conjugated secondary antibody. Specific bands were visualized using an amplified Opti-4CN substrate kit. NP40-soluble fractions incubated with anti-GFAP PAb were further incubated with sheep anti-rabbit IgG in blocking buffer followed by peroxidase–anti-peroxidase complex in TBS–Tween. DAB-HCl and H2O2 in 100 mM Tris-HCl, pH 7.4, were used as substrates. NP40-soluble fractions incubated with anti-GFAP MAb were further incubated with HRP-conjugated rabbit anti-mouse IgG and the reaction was developed using DAB-HCl and H2O2 in 100 mM Tris-HCl, pH 7.4, as substrates.
LM Immunocytochemistry
Small pancreatic, intestinal, and brain samples, as well as aliquots of purified islets, were fixed in 4% paraformaldehyde in 0.1 M cacodylate buffer (pH 7.4) for 3 hr at 4C and processed for routine embedding in Epon 812. Four pancreata and brains were fixed for 12–16 hr in 10% formalin or Bouin's fluid and treated for standard embedding in paraffin.
Sections (~5–6 μm thick) from paraffin-embedded samples were mounted on poly-
Consecutive semithin sections (~0.7 μm thick) from Epon-embedded samples of pancreas and intestine were mounted on poly-
Several immunocytochemical control experiments were carried out to substantiate GFAP PAb specificity: omission of the primary antibody or its replacement with normal rabbit serum, liquid-phase absorption with poly-
Immunoelectron Microscopy
Small samples of pancreatic and brain tissues were fixed in 4% paraformaldehyde in 0.01 M PBS (pH 7.4) for 3 hr at 4C and processed for routine embedding in Lowicryl resin. Thin sections were mounted on formvar-coated nickel grids. Grids were incubated on 1% BSA in PBS for 30 min at RT and then placed on a drop of 1:10 anti-GFAP PAb in PBS overnight at 4C. Then the grids were rinsed with PBS, incubated with 1% BSA in PBS, and placed on a drop of 1:20 20-nm gold-conjugated anti-rabbit IgG in PBS for 90 min at RT. After several rinsings with PBS and distilled water, grids were dried, stained with uranyl acetate, and observed with a Philips 201 transmission electron microscope.
Results
GFAP Is Expressed in Pancreatic Glucagon-secreting Cells
To minimize the risk of crossreaction with other IFs, immunocytochemical detection of GFAP-immunoreactive cells in mouse pancreas was performed with two commercially available anti-GFAP PAbs raised using different sources of immunogen (human brain and bovine spinal cord). Both antibodies gave essentially the same results: acinar and duct cells as well as blood vessels were completely unstained. In contrast, Schwann cells of major nerve fibers were slightly GFAP-immunoreactive. In addition, both PAbs stained cells preferentially located in the periphery of almost all islets of Langerhans (Figure 1). To identify which type of islet cell was GFAP-immunoreactive, we carried out experiments staining three consecutive semithin sections (0.7 μm thick) with anti-somatostatin, anti-GFAP, and anti-glucagon antisera. Whereas no somatostatin-secreting cells were GFAP-immunoreactive (Figures 2A and 2B), all glucagon-secreting cells stained with the anti-GFAP PAb (Figures 2B and 2C).
To verify whether the antibodies employed in this study recognized actual GFAP, we purified the islets of Langerhans and carried out Western blotting analysis of their IF-enriched cytoskeletal fraction. The same protocol was followed with mouse brain samples as a positive GFAP-immunoreactive control. The enzymatic digestion of the pancreas and its subsequent centrifugation through a discontinuous density gradient (see Materials and Methods) resulted in efficient isolation of islets of Langerhans, which was further improved by hand-picking the islets. This procedure is presumed to remove nerve fibers and periacinar stellate cells that might contaminate the sample with GFAP. To determine if any contamination had occurred during the preparation, aliquots of purified islets were immunocytochemically tested to detect GFAP-containing cells. All the experiments performed revealed GFAP immunoreactivity exclusively in glucagon-secreting cells (Figure 3). No other cells, among the few elements contaminating the isolated islets, stained with anti-GFAP Abs. Western blotting analysis with anti-GFAP PAbs of IF-enriched cytoskeletal fractions revealed a single band in pancreatic samples co-migrating at 50 kD with immunoreactive GFAP from brain tissue (Figure 4B). As an additional control to confirm that the 50-kD band was GFAP, we performed a new series of experiments by immunoblotting membranes with an anti-GFAP MAb (clone 6F-2). In this case, however, whereas brain samples displayed, as expected, a single band with a relative mobility corresponding to 50 kD (Figure 4C), pancreatic samples exhibited two distinguishable GFAP-immunoreactive bands of similar molecular weights (50 and 51 kD).
GFAP Is Present Mainly in Its NP40-soluble Form
Taken together, these results are consistent with the expression of GFAP in glucagon-secreting cells. However, the amount of GFAP belonging to the cytoskeletal fraction appeared very low because we had to use an amplified Opti-4CN substrate kit to detect it. Because of this, we decided to verify whether the immunocytochemical signal could also be due to a soluble antigen present in the cytosol of glucagon-secreting cells. For this purpose, Western blotting analysis with anti-GFAP PAbs was conducted on NP40-soluble fractions of islets and brain, revealing a single 50-kD band in both pancreatic and brain samples (Figure 5a). To confirm that the 50-kD band of both samples really corresponded to GFAP, we carried out the same controls performed for the IF-enriched cytoskeletal fractions, incubating membranes with an anti-GFAP MAb (clone 6F-2). Western blotting analysis with the latter Ab revealed only one 50-kD band in pancreatic samples (Figure 5B), whereas no bands were detectable in brain samples. On the basis of these results, we can affirm that GFAP is expressed in glucagon-secreting cells as either polymerized or soluble forms.
Even though we did not directly quantify these two pools of GFAP, the ratio between polymerized GFAP and its detergent-soluble form can be roughly assessed by calculating the dilution factors of the respective samples loaded onto the gels. The GFAP NP40-soluble pool was diluted about 400 times compared to its polymerized form before being loaded onto the gel. Moreover, despite this greater dilution, the demonstration of GFAP-immunoreactive bands in the NP40-soluble sample did not require any particularly sensitive detection device, as did the IF-enriched cytoskeletal fraction, which needed an amplified detection system (Figures 4 and 5).
Taken together, these findings show that in glucagon-secreting cells the GFAP NP40-soluble pool appears to be a hundred times more highly represented than its polymerized form.
GFAP Is Associated with Secretory Granules
To localize GFAP in glucagon-secreting cells, we carried out an immunoelectron microscopic analysis of GFAP subcellular distribution. As a positive control, sections from mouse brain were also handled with the same immunocytochemical protocol. Accordingly, in the latter case, gold particles selectively labeled bundles of IFs located in astrocyte processes (Figure 6).
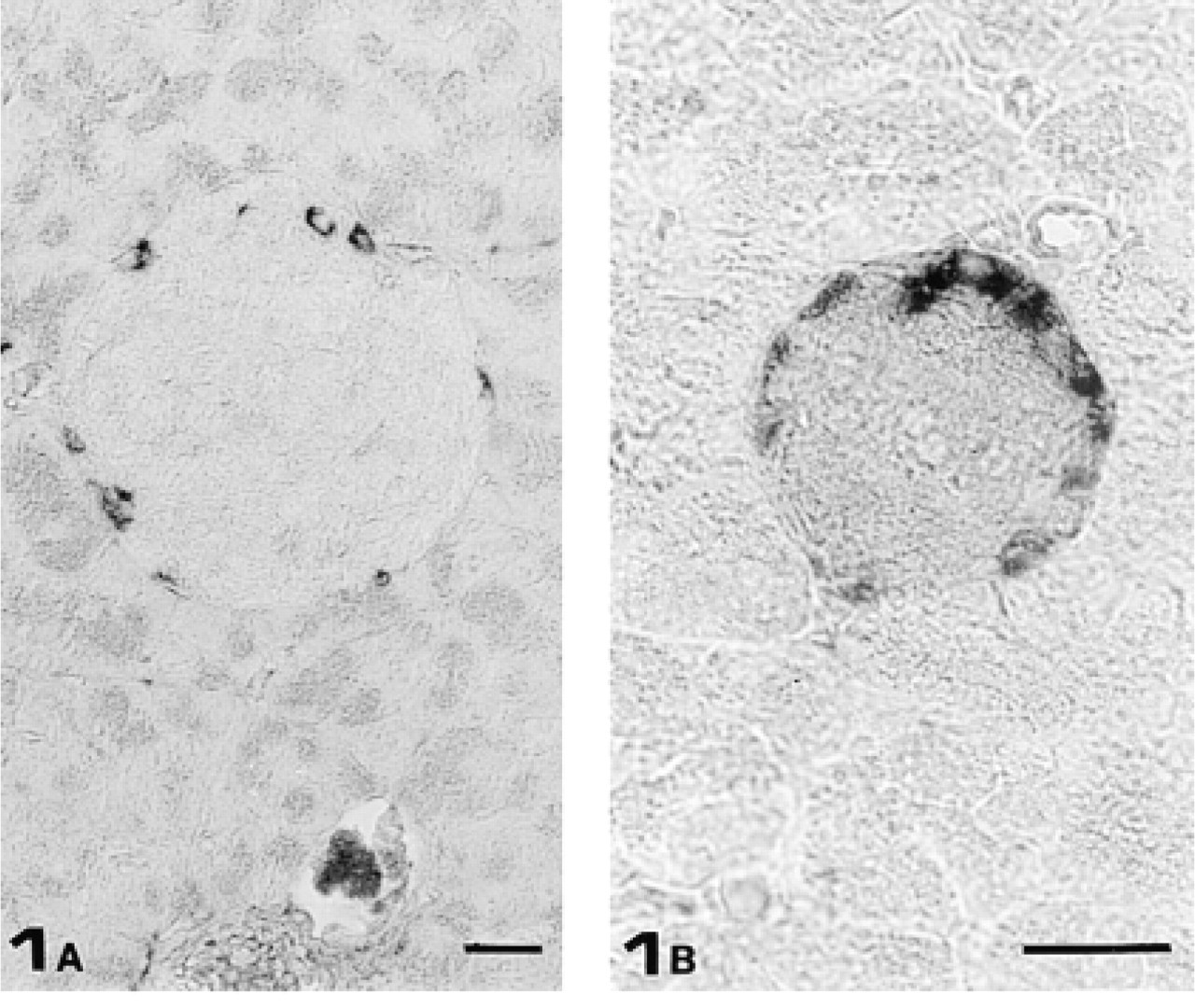
(
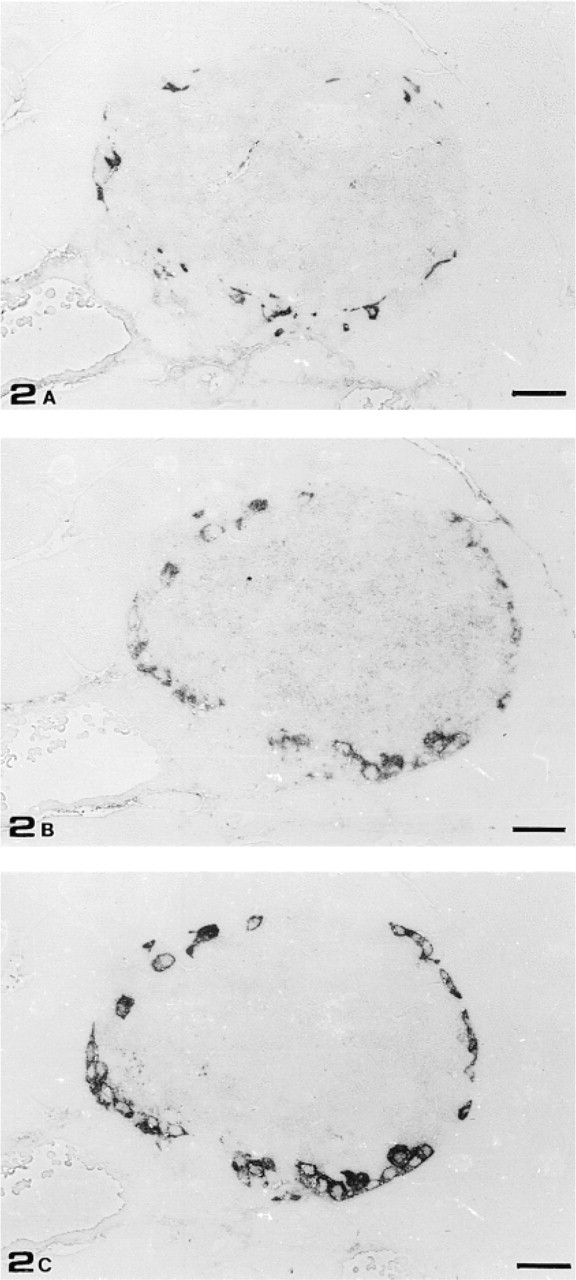
Consecutive semithin sections of mouse islet of Langerhans stained with anti-somatostatin PAb (
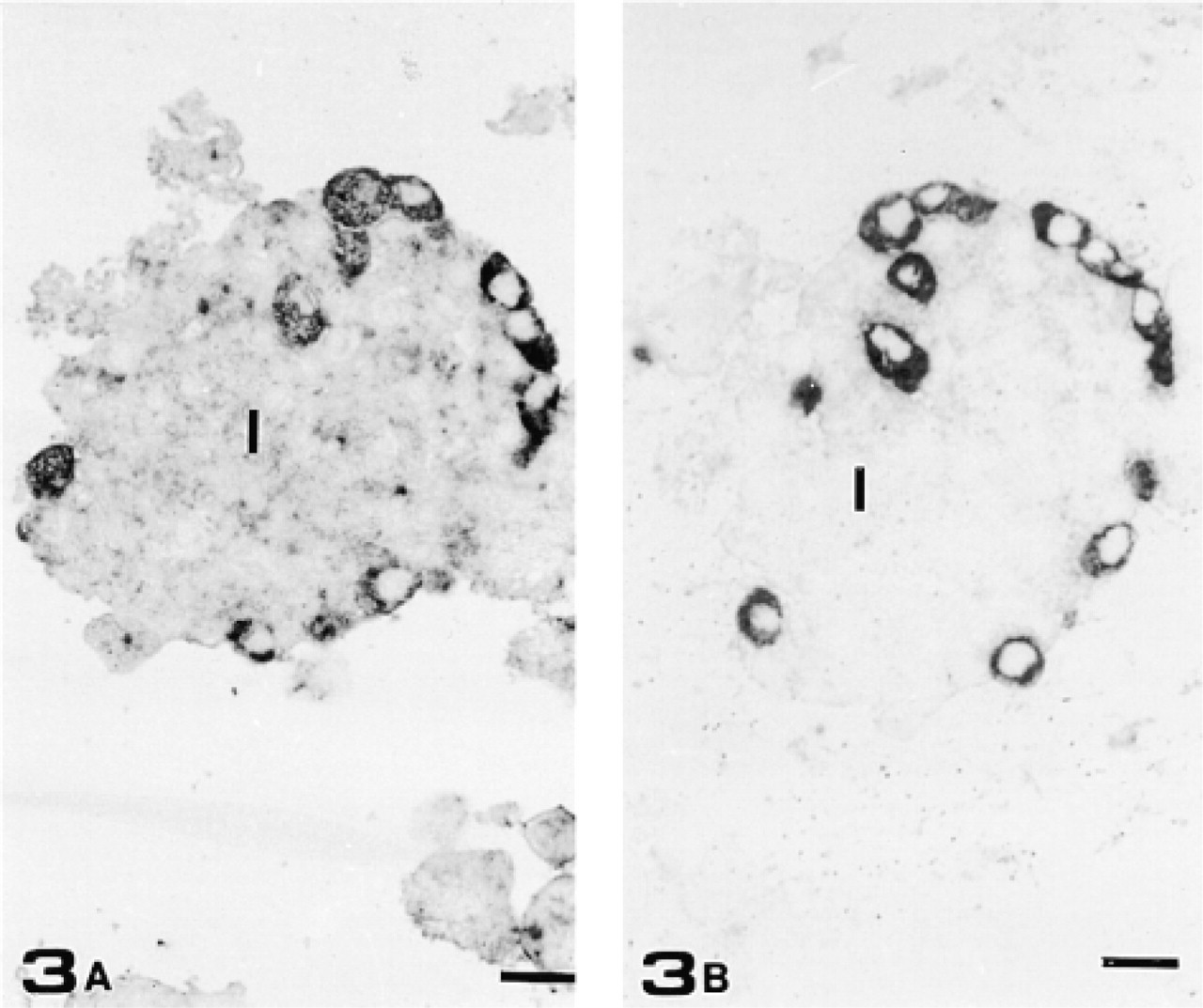
Consecutive semithin sections of purified islets stained with anti-GFAP PAb (
On pancreatic sections, confirming the LM findings, the reaction was restricted to glucagon-secreting cells (Figure 7A). In this case, consistent with biochemical analysis that referred most of the GFAP immunoreactivity to the NP40-soluble cell fraction, gold particles did not label filamentous structures. Surprisingly, however, gold particles were not scattered throughout the cytoplasm as would be expected but were associated with the periphery of secretory granules (Figure 7B).
Immunocytochemical Controls
Light microscopic control experiments consisting of the omission of anti-GFAP PAb or its replacement with normal rabbit serum abolished specific staining on both brain and pancreatic sections (data not shown).
Because unspecific binding of immunoglobulins to basic peptides has been reported to occur in glucagon-secreting cells as well (Buffa et al. 1979), immunocytochemical experiments, resulting in substantially unaltered staining, were carried out after overnight anti-GFAP PAb liquid-phase absorption with poly-
Anti-GFAP PAb liquid-phase absorption with purified GFAP abolished specific staining on both brain and pancreatic sections (Figures 8A and 8B). Positive control experiments were carried out on brain and intestine sections, resulting in staining of scattered brain astrocytes and satellite cells of intestinal myoenteric and submucosal plexi (Figures 8C and 8D).
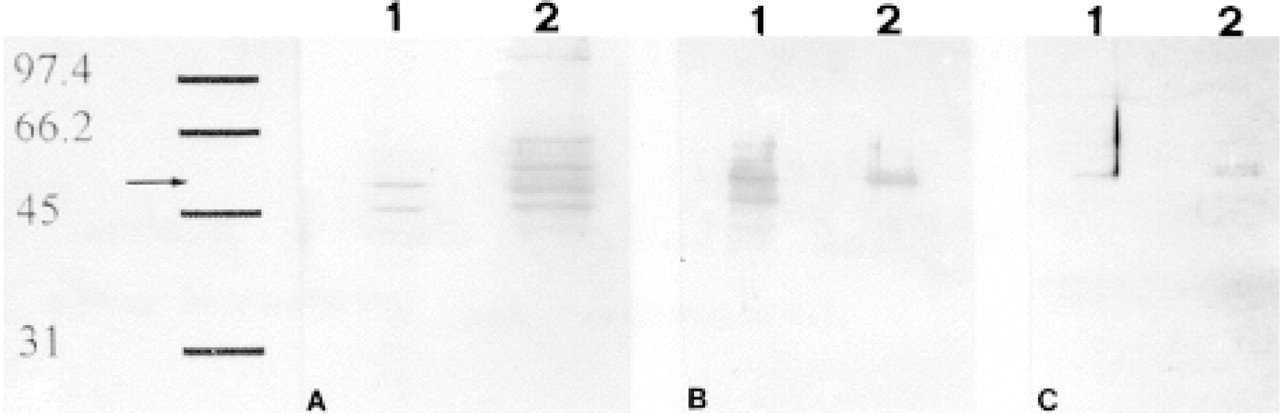
IF-enriched cytoskeletal fractions of brain and islet-enriched pancreatic tissues. (
Discussion
In this study we describe the expression of GFAP in glucagon-secreting cells of adult mouse pancreas. This finding confirms previous observations of GFAP-like immunoreactivity in glucagon-secreting cells of rat pancreas (Regoli et al. 2000) and, corroborated by combined immunocytochemical and biochemical investigations with a panel of three anti-GFAP Abs, supports the presence of actual GFAP in glucagon-secreting cells. This finding has particular relevance because that the IFs thus far reported to be stably expressed in pancreatic epithelium belong exclusively to the CK (Types I and II) subfamily. Moreover, in vivo co-expression of CKs and vimentin-like IFs is an extremely rare phenomenon in non-transformed endoderm-derived cells of normal adult animals. This feature, to our knowledge, has been described with certainty exclusively in a subpopulation of cells from salivary glands (Achtstätter et al. 1986b).
More important than the simple finding of its presence in glucagon-secreting cells is the demonstration that GFAP is mostly expressed in its non-polymerized form (i.e., NP40-soluble fraction). This raises basic questions about the role of IFps. Even though additional functions for IFps have been strongly suspected owing to their dynamic nature (Klymkowsky 1995; Liao and Omary 1996; Chou et al. 1997), in most cases IFps have been considered merely as a sort of building block destined to make up IFs, and questions about their function have been relegated to the role played by IFs themselves. If this were the case for GFAP in glucagon-secreting cells as well, we would assume that such a large detergent-soluble pool should exist as a reservoir of molecules ready to polymerize quickly under precise conditions. However, this view is not supported by any evidence for the existence of such a mechanism in other cell types. IFps in vivo have been previously found largely in their polymerized state, and a detergent-soluble pool is often reported to be barely detectable (Blikstad and Lazarides 1983; McTavish et al. 1983; Moon and Lazarides 1983; Soellner et al. 1985; Isaacs et al. 1989). The extent of this pool has been determined to vary according to the type of cell and the type of IF. For example, soluble CK8/18 tetramers have been evaluated as 1–10% of the total CK8/18 content in Xenopus laevis oocytes (Gall and Karsenti 1987) and as 4–6% of the total content in the human colon epithelial cell line HT29 (Chou et al. 1993). Even less represented, soluble vimentin has been assessed as ranging between 0.18 and 0.39% of the total vimentin content in cultured rat cells (Soellner et al. 1985). These estimations are generally in accordance with a model of IFs predicting a dynamic equilibrium with a soluble pool of IFps. We showed, however, that in glucagon-secreting cells the amount of the GFAP NP40-soluble pool appears to be two orders of magnitude greater than its polymerized form, as if GFAP subunits had functions alternative to those of forming polymerized IFs. Thus far, a certain number of mechanisms have been described to influence IFp solubility, but it is not clear if the maintainence of such a large soluble pool can be achieved by the same molecular mechanisms. The first mechanism previously reported involves post-translational modifications of IFps by serine phosphorylation (Steinert and Liem 1990; Fuchs and Weber 1994), which appears to be proportional to the degree of solubility (Liao and Omary 1996). In this regard, the detection with an anti-GFAP MAb of two well-distinct immunoreactive bands with very close molecular weight in the cytoskeletal fraction of pancreatic samples likely discloses the existence of post-translational processes. However, not all the post-translational modifications of IFps appear to play a significant role in modifying IFp solubility, as in the case of threonine/serine glycosylation (Chou et al. 1993). A second mechanism affecting IFp solubility is the association with other proteins that may act as solubility co-factors (Chou et al. 1993). One example is the 14-3-3 protein, which has been reported to associate with phosphorylated CK8/18, enhancing its solubility (Liao and Omary 1996). We are now engaged in determining whether NP40-soluble GFAP is associated with other cytoplasmic proteins.
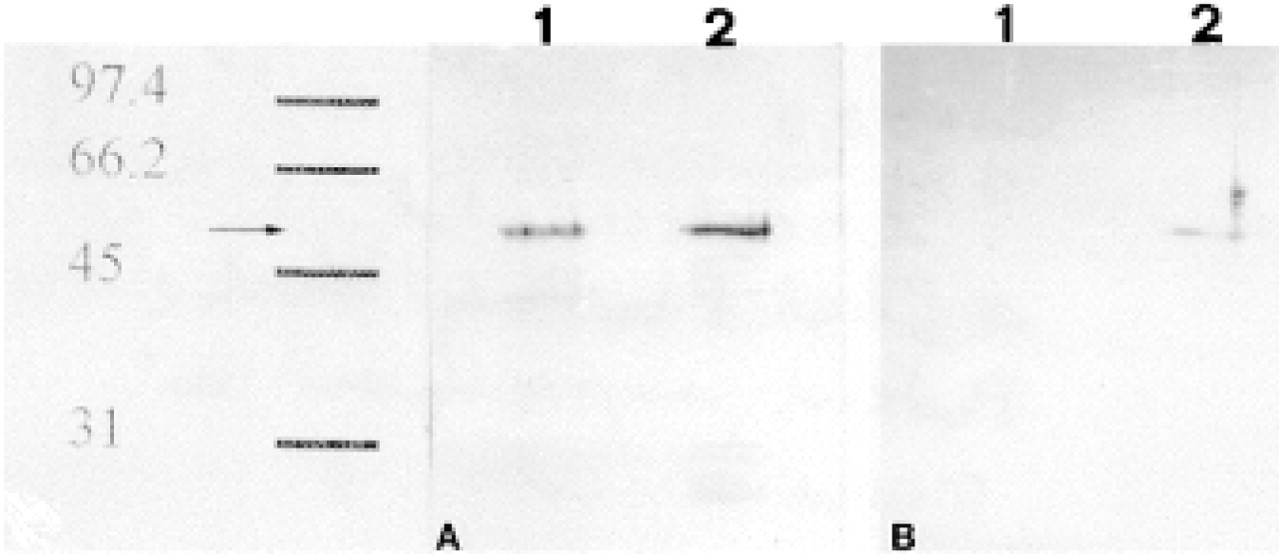
NP40-soluble fractions of brain tissue and purified islet of Langerhans. (
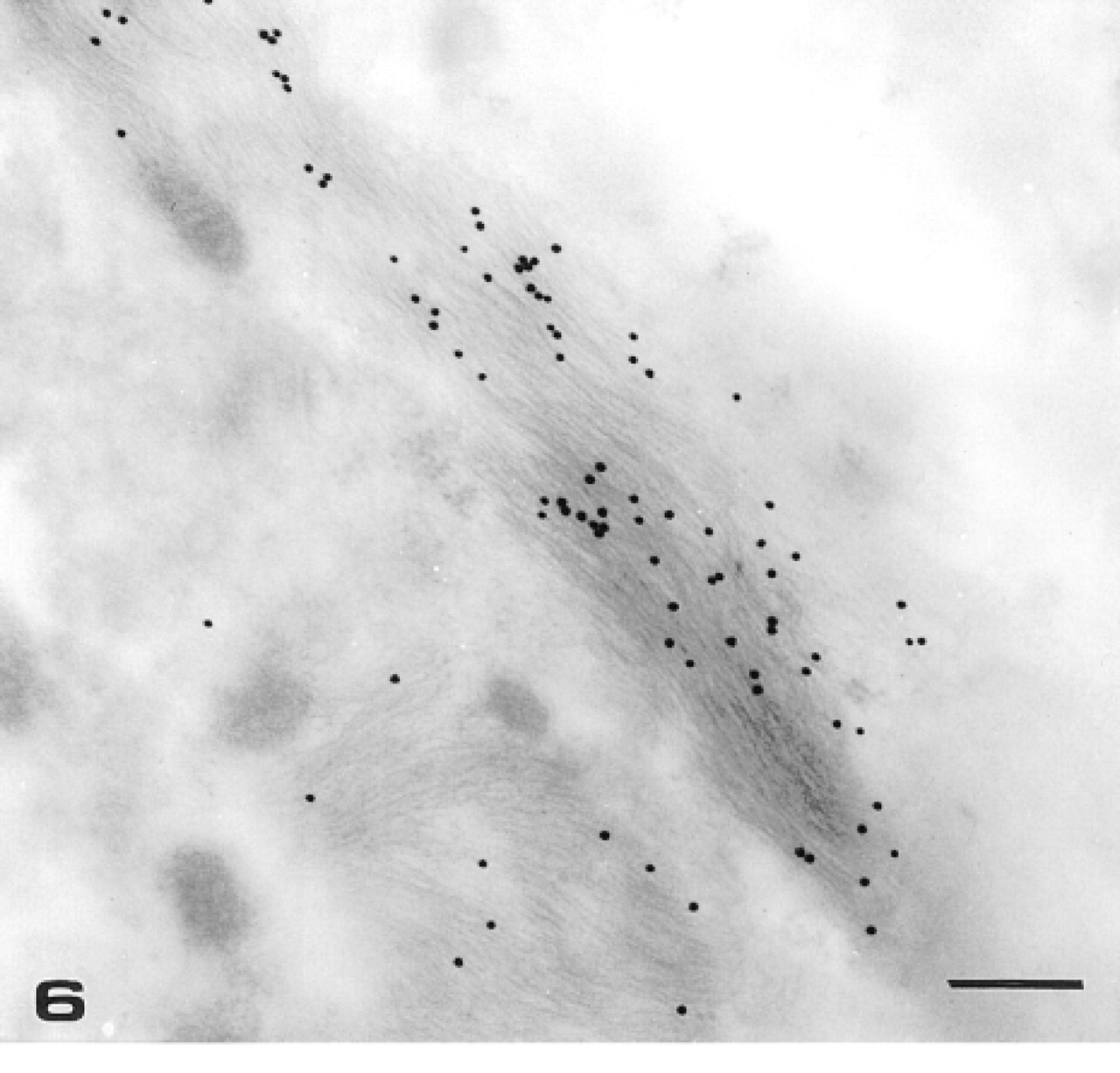
TEM micrograph of mouse brain. Detail of an astrocyte process immunolabeled with anti-GFAP PAb. Twenty-nm gold particles selectively label bundles of intermediate filaments. Bar = 320 nm.
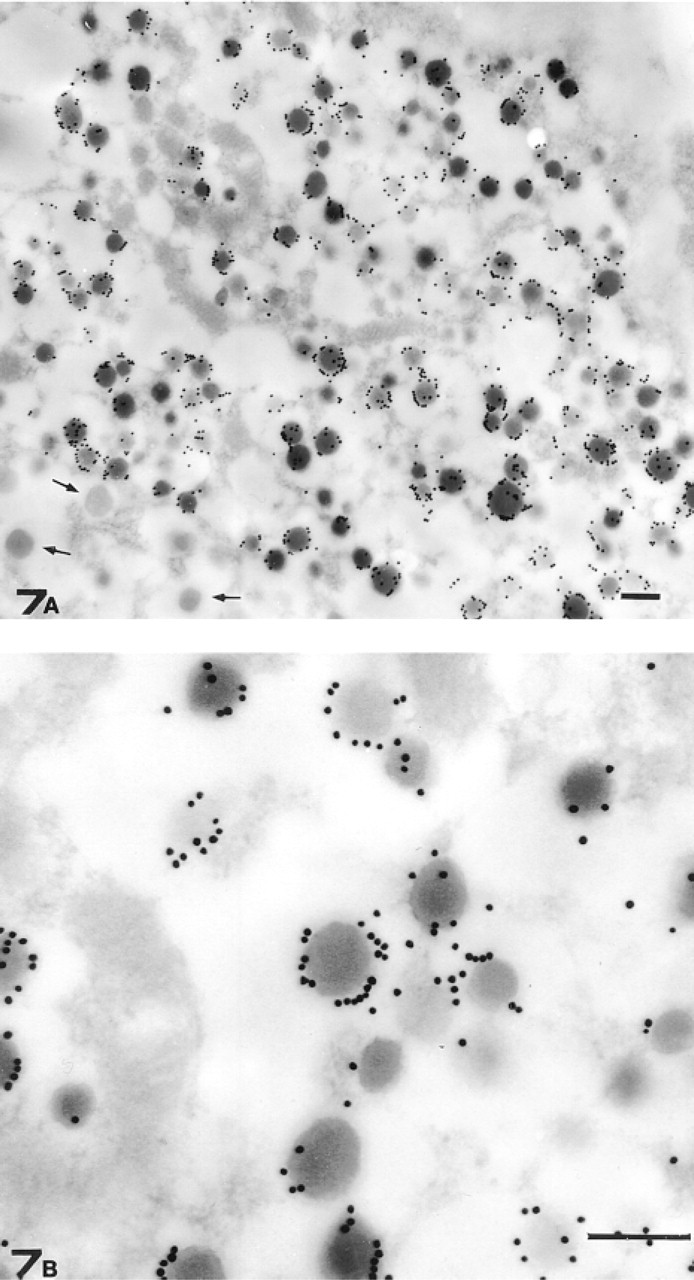
TEM micrograph of a mouse islet of Langerhans. (
At present it is not clear which cellular roles such large amounts of soluble GFAP could play. However, our immunoelectron microscopic analysis of subcellular GFAP distribution in glucagon-secreting cells, sheds some light that enables us to formulate concrete hypotheses. Instead of labeling filamentous structures as occurs in brain astrocytes, in glucagon-secreting cells gold particles localize to the periphery of secretory granules, corroborating previous conjectures about an involvement of GFAP in secretion (Regoli et al. 2000). Other cytoskeletal elements have been previously reported to associate with secretory granules such as actin, myosin, and even keratin (Bendayan 1981, 1985; Bendayan et al. 1982). Whereas the involvement of myosin (and associated actin filaments) in membrane trafficking and in secretion has been recently reproposed (Poucell–Hatton et al. 1997; Wu et al. 2000), keratin detection on the periphery of pancreatic zymogen granules (Bendayan 1985) could acquire new importance in view of a more generalized involvement of IFps in secretion.
The association of GFAP with the periphery of secretory granules could be due to direct connections with the limiting membrane of granules as the result of IFp interactions with membrane lipids, as previously suggested (Gyoeva and Gelfand 1991), or of binding to associated proteins. The ability of vimentin to associate directly with lipids has been demonstrated in vitro and, in this context, the observation that such an association inhibits IF assembly appears particularly interesting (Perides et al. 1987; Traub et al. 1987). Moreover, noncovalent binding of lipids to vimentin and keratin has been described after extraction of cells with detergent (Asch et al. 1990), and a membrane-associated pool of CK8/18 tetramers has been recently hypothesized (Liao and Omary 1996).
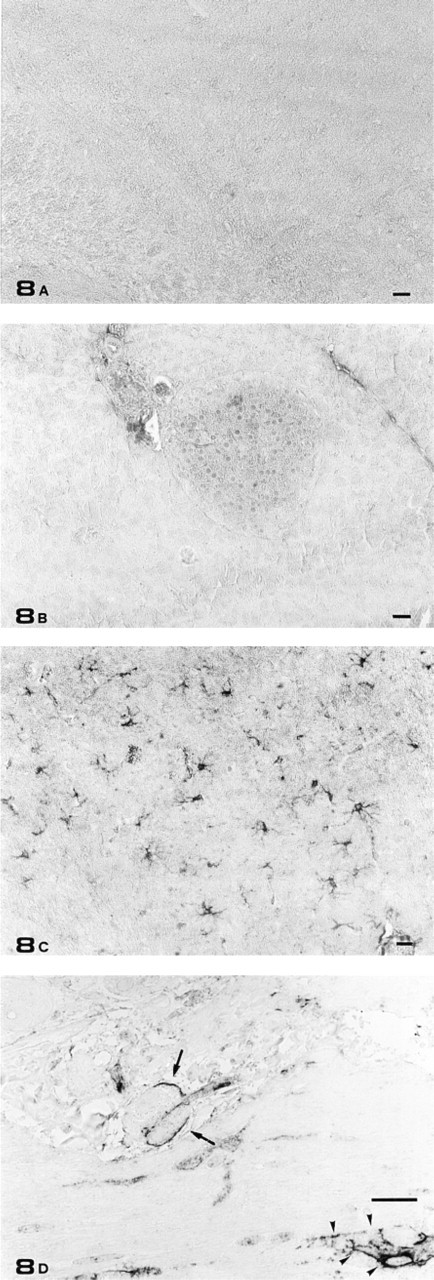
Light microscopic immunostaining controls. (
Despite these observations, GFAP interaction with secretory granules may be more likely mediated by associated proteins. Many proteins have been recently described as being associated or as being capable of associating with IFs or with their subunits, thus potentially modulating their role. For example, 14-3-3 protein, in addition to associating with CK8/18 tetramers, has been described to be involved in priming regulated exocytosis in permeabilized chromaffin cells (Chamberlain et al. 1995), thus representing a possible prototype of molecules that could link together IFps, signal transduction events, and secretory activities.
However, other proteins described as associating with IFs are also involved in secretion, as in the case of kinesin. Kinesin, a plus-end microtubule-directed motor protein, in addition to binding microtubules, associates with IFs, thus interacting directly or indirectly with IFps (Gyoeva and Gelfand 1991; Liao and Gundersen 1998; Prahlad et al. 1998). Kinesin involvement in Ca2+-regulated exocytotic events has been recently reported (Bi et al. 1997), and its implication in secretion is further supported by the demonstration that regulated secretion is heavily impaired in pancreatic β-cells and acinar cells by the respective microinjection of kinesin heavy chain antisense oligonucleotides and anti-kinesin Abs (Meng et al. 1997; Marlowe et al. 1998). Therefore, the localization of GFAP around glucagon granules could take part in a cross-bridging system linking microtubules and kinesin on one side and glucagon granule membrane on the other side.
Whatever the role of GFAP is, its selective expression in glucagon-secreting cells raises a basic question for islet cell biology that will require additional investigation. Is the location of soluble IFps in relation to secretory granules a unique feature that distinguishes glucagon-secreting cells from the other endocrine cell types? In this case, we should assume that GFAP modulates an exclusive function most likely related to specific secretory products of glucagon-secreting cells. Otherwise, if GFAP plays a role in glucagon-secreting cells that is common to all islet cells, it is reasonable to predict the expression of other molecules (maybe still IFps) replacing GFAP in the other pancreatic endocrine cell types.
Footnotes
Acknowledgements
Supported by local funds from the University of Siena (GL and AB).
We wish to thank Mr P. Salvatici and Ms D. Orazioli for excellent technical assistance. We are grateful to Dr A. Gugliucci for critical revision of the manuscript and to Dr M. Bendayan for fruitful discussions during the preparation of the manuscript. We are indebted to Dr L. M. Keith for improving the English text.