
Abstract
Keywords
In diagnostic cytology of the uterine cervix, cytomorphological evaluation of Papanicolaou-stained smear preparations (Pap smears; Papanicolaou and Bridges 1942) is the gold standard. Invasive cervical cancer can be diagnosed and cellular changes can be identified at an early dysplastic stage, so that women can be treated before severe abnormalities occur. National population screening programs in Europe and North America have contributed significantly to reduction of the incidence and mortality of cervical cancer. However, false-negative and false-positive results have limited the value of the Pap test. Consequently, Pap smear examination may lead to substantial under- or overtreatment of women.
To improve the reliability of diagnostic cytology, molecular techniques can be used to detect non-morphological markers as an adjunct to the conventional cytomorphology. Potential molecular biomarkers of cervical intraepithelial lesions are now under investigation and await implementation in routine diagnostic procedures. Candidate ancillary tests include monitoring for Ki-67 and p53 expression with immunocytochemistry (Holm et al. 1993; Bulten et al. 1996; Isacson et al. 1996), numerical chromosomal aberrations with in situ hybridization (Segers et al. 1994; Bulten et al. 1998), nuclear DNA index and chromatin texture features with image cytometry (Hanselaar et al. 1998), and HPV presence and typing with the polymerase chain reaction (PCR; Walboomers et al. 1994; Ramael et al. 1995).
Additional molecular techniques can be applied only if a sufficient number of preparations are made from a single cell sample. In present day practice, however, only a single smear is made for routine examination. Valuable diagnostic cells present on the brush are discarded but might be used for the preparation of extra microscopic specimens (Goodman and Hutchinson 1996). Strategies to prepare multiple cytological specimens from one cell scraping include mono-layer and cell block techniques. Monolayer preparation by cytospin or the new-generation monolayer systems, such as ThinPrep and AutoCyte, allow the preparation of three to five diagnostic samples from each scraping. Cell block techniques, by which cytological samples are embedded in paraffin, can yield up to 20-50 sections per scraping. Protocols of the latter include direct embedding of a cell pellet in paraffin (Krogerus and Andersson 1988; Domagala et al. 1990) or indirect embedding via uptake of the cells in agar (Olson et al. 1986; Kung et al. 1990) followed by processing of the cell clump into paraffin. To date, the use of cell block preparations has been limited to cytomorphological screening of fine-needle aspirates or cell effusions and the application of a single additional test, such as immunoperoxidase staining, in situ hybridization, or PCR (O'Brien et al. 1980; Unger et al. 1989; Chen et al. 1993).
The aim of this study was to develop a cell block processing procedure (AgarCyto) for brush material of the uterine cervix that would enable us to assess cytomorphology and to perform multiple ancillary molecular techniques using standard laboratory protocols. Here we describe the optimization of fixation and cell embedding concerning cytomorphology and signal-to-noise ratio of immunocytochemical staining and in situ hybridization techniques. Furthermore, we describe the use of AgarCyto sections for HPV typing by line probe assay (LiPA)-PCR and an additional step in the protocol for DNA index measurements by image cytometry.
Materials and Methods
Cell Culture and Cytological Specimens
For the development of the AgarCyto cell block procedure and the protocols for ancillary molecular techniques, five different cell lines were used. Human cervical carcinoma cell lines HeLa, SiHa, and CaSki, human lymphoblast leukemia cell line Molt-4, and human vulval carcinoma cell line A431 were used for optimization of immunocytochemistry (ICC), in situ hybridization (ISH), and PCR. A431 and Molt-4 were also used for optimization of DNA image cytometry. All cell lines were obtained from the American Tissue and Cell Collection (ATCC; Manassas, VA) and were cultured according to the supplier's instructions. For validation of the techniques on clinical samples, 50 cervical scrapings were collected at the Department of Obstetrics & Gynecology (University Hospital Nijmegen, The Netherlands).
Fixations
To optimize for a good balance between cytomorphology and signal-to-noise ratios of ICC and ISH, chessboard experiments were performed in which fixation types and times were varied (1). Subconfluently grown cell cultures were trypsinized and fixed in suspension for 2, 16, and 64 hr in one of the following fixatives: (a) 50% ethanol/2% carbowax, a standard collection fluid in cytology (Bibbo 1991); (b) 50% ethanol/2% carbowax/4% formaldehyde, a fixative that combines cytological and histological features; (c) CytoRich (Roche; Elon College, NC), an ethanol-based fixative developed for use in monolayer preparations (McGoogan and Reith 1996); (d) Neutral-buffered 4% formaldehyde, a standard fixative in histology (Pearse 1980); and (e) Unifix (Klinipath; Duiven, The Netherlands), a fixative containing formaldehyde, zinc sulfate, and other constituents, which has shown to give excellent immunohistochemical results (Mugnaini and Dahl 1983; Dapson 1993). After fixation, the cell suspensions were processed into AgarCytos.
Clinical material was obtained using a Cervex brush (Rovers; Oss, The Netherlands), and a routine cervical smear was made. The brush with remnant material was put into a storage vial with primary fixative and transported (Figure 1A). Within 16 hr, cervical cells were transferred from the transport vial into 50-ml tubes (Figure 1B), spun down at 2000 rpm for 10 min, secondarily fixed in suspension for up to 64 hr, and processed into AgarCytos.
AgarCyto Cell Block Preparation
The AgarCyto cell block preparation is based on earlier protocols described by Olson et al. (1986) and Kung et al. (1990), who used agar as an intermediate embedding medium. We improved their methods with respect to cell yield and applicability to multiple molecular diagnostic analyses. Fixed cells were pelleted by centrifugation for 10 min at 2000 rpm. The supernatant was discarded and the pellet was carefully loosened, transferred into a 1.5-ml Eppendorf reaction tube, and spun down for 10 min at 2000 rpm. The pellet was resuspended in 1 ml 2% liquid agarose at 65C (LE, analytical grade; Promega, Madison, WI). Again, the reaction tube was centrifuged for 5 min at 1000 rpm to concentrate the cells in the agar (Figure 1C). The agar-cell pellet was solidified at 4C for at least 1 hr. The agar cone was carefully taken out of the reaction tube with pointed forceps and laterally divided in two halves (Figure 1D). The two agar pieces were wrapped in filter paper for biopsy preparation (Medite; Burgdorf, Germany), placed in a Tissue-Tek cassette (Sakura FineTek; Zoeterwoude, The Netherlands), and embedded in paraffin using an automated tissue processor (Tissue-Tek VIP150; Sakura) under standard conditions for surgical biopsies. A paraffin embedded agar-cell pellet is called AgarCyto (Figure 1E). From the AgarCyto, 4-± sections were cut, mounted on SuperFrost/Plus glass slides (Menzel-Gläser, Germany), and dried overnight at 56C. For cytomorphological examination, AgarCyto sections were stained with hematoxylin-eosin (H&E).
Immunocytochemistry
ICC detection for Ki-67 and p53 in AgarCytos of tumor cell lines and cervical scrapings was demonstrated by standard immunocytochemical procedures. Four-±-thick sections were dewaxed in xylol and methanol and endogenous peroxidase was blocked for 15 min in 1% H2O2 in methanol. Antigens were unmasked by microwave heating in buffered 0.01 M citrate solution, pH 6.0, for 3 min at 850 W and 10 min at 150 W, and cooled to room temperature (RT) in the same solution for 30 min. Preparations were rinsed in PBS for 20 min. All primary antibodies were diluted in PBS and incubated overnight at 4C, and all following antibodies were diluted in PBS/1% bovine serum albumin (BSA; Sigma, St Louis, MO) and incubated for 30 min at RT. All washings between the incubations were performed in PBS. For p53 immunodetection, slides were preincubated with 10% normal horse serum in PBS and then incubated with DO-7 monoclonal antibody (MAb) against wild-type and mutant p53 (1:50; Novocastra, Newcastle, UK), biotinylated horse anti-mouse (1:200; Vector Laboratories, Burlingame, CA), and horseradish peroxidase avidin-biotin complex (ABC Elite; Vector Laboratories). Ki-67 was detected, without preincubation, by MAb Mib-1 (1:50; Dianova, Hamburg, Germany), followed by horseradish peroxidase-conjugated rabbit anti-mouse (1:100; DAKO, Glostrup, Denmark). Peroxidase was visualized with 0.05% diaminobenzidine (DAB; Sigma)/0.15% H2O2 in PBS for 5 min at RT. Specimens were counterstained with hematoxylin and mounted in Permount (Fisher Scientific; Fair Lawn, NJ).
Influence of fixation on morphology, immunocytochemistry, in situ hybridization, and LiPA-PCR in tumor cell lines
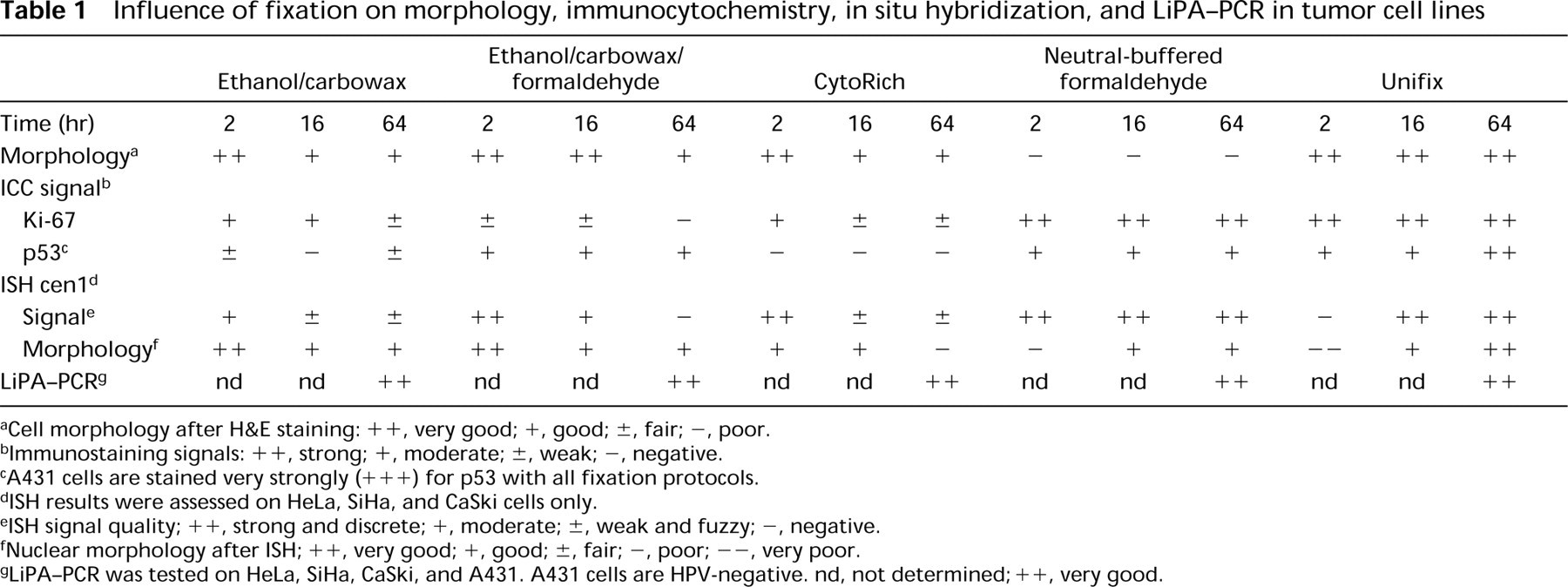
aCell morphology after H&E staining: ++, very good; +, good; ±, fair; -, poor.
bImmunostaining signals: ++, strong; +, moderate; ±, weak; -, negative.
cA431 cells are stained very strongly (+++) for p53 with all fixation protocols.
dISH results were assessed on HeLa, SiHa, and CaSki cells only.
eISH signal quality; ++, strong and discrete; +, moderate; ±, weak and fuzzy; -, negative.
fNuclear morphology after ISH; ++, very good; +, good; ±, fair; -, poor; -, very poor.
gLiPA-PCR was tested on HeLa, SiHa, CaSki, and A431. A431 cells are HPV-negative. nd, not determined; + +, very good.
In Situ Hybridization
DNA probe pUC1.77 for the centromere region of chromosome 1 (cen 1; Cooke and Hindley 1979) and genomic DNA probe specific for human papilloma virus Type 16 (HPV 16) were labeled by nick-translation with biotin-16-dUTP according to the supplier's instructions (Boehringer; Mannheim, Germany). For both probes, the hybridization protocol applied to AgarCyto sections was essentially as described (Kerstens et al. 1995). AgarCyto sections were consecutively dewaxed, blocked for endogenous peroxidase, and pretreated with 1 M NaSCN for 10 min at 80C. Protein digestion was performed using 4000 U/ml pepsin (Sigma) in 0.2 M HCl for 5 min at 37C, and sections were dehydrated through an alcohol series and air-dried. Two ng/μl DNA probe was dissolved in 15 μl hybridization mix containing 60% formamide, 2 × standard saline citrate (SSC), pH 7.0, 10% dextran sulfate (Sigma), and 50 ng/μl herring sperm DNA (Boehringer). The probe mix was applied to the sections, covered with a coverslip, and sealed with rubber cement. Probe and target DNA were heat-denatured simultaneously for 10 min at 80C and hybridized overnight at 37C in a moist chamber. Coverslips were removed by immersing the slides in 2 × SSC, pH 7.0, at 42C. Posthybridization washes were carried out at 42C in 60% formamide/2 × SSC, pH 7.0, twice for 5 min and in 2 × SSC, pH 7.0, twice for 5 min. The slides were rinsed in PBS/0.05% Tween-20 (PBST) and the biotinylated probe-target hybrid was detected by mouse anti-biotin (1:100; DAKO), biotinylated horse anti-mouse, and peroxidase-ABC as described for immunocytochemistry.
For detection of HPV 16 DNA, the catalyzed reporter deposition (CARD) signal amplification method was used (Hopman et al. 1998). The hybridized samples were subsequently detected with horseradish peroxidase-conjugated avidin (1:100) in PBST for 30 min at 37C, biotinylated tyramide (1:100) in PBS/imidazol/0.001% H2O2 for 5 min at RT, and horseradish peroxidase ABC (1:100) for 15 min at 37C. Peroxidase was visualized with DAB as described for ICC.
Microscopic Evaluation
To assess the quality of the experimental immunostaining, the results were correlated to Ki-67 and p53 immunoexpression levels of these cell lines as known from the literature. Ki-67 antigen is expressed in proliferating cells and is therefore present in more than 50% of cells of a subconfluent cell culture (Sasaki et al. 1989; Benbrook et al. 1995; Dong et al. 1997). For development of sensitive p53 immunostaining, we used (a) HPV-containing cervical cancer cell lines expressing low levels of wild-type p53 due to degradation by HPV E6 antigen (Scheffner et al. 1991; Wrede et al. 1991; Liang et al. 1993) and (b) Molt-4 cells expressing very low levels of mutant p53 (Chow et al. 1993). As a positive control A431 cells were used, which stongly express mutant p53 (Reiss et al. 1992). For quality assessment of the ISH experiments, the number and intensity of ISH signals were correlated to the copy number of cen 1 and approximate copy number of HPV 16 genomes and number of integration sites as described in the literature. Classical and molecular cyto-genetic studies of HeLa have shown four copies of cen 1, five integration sites for HPV 18, and none for HPV 16 (Popescu and DiPaolo 1989; Macville et al. 1999). SiHa contains three copies of cen 1 and one copy of HPV16 at one integration site (Mincheva et al. 1987; Popescu and DiPaolo 1989). CaSki contains 200-500 copies of HPV 16 at 11 integration sites, is hypertriploid, and contains three or more copies of cen 1 (Mincheva et al. 1987). HeLa and A431 cells were used as negative controls for HPV 16 ISH. The influence of the different fixation procedures on cell morphology, ICC, and ISH staining efficiency and signal-to-noise ratios were qualitatively scored. The optimized protocol for cell lines was validated on cervical scrapings.

Preparation of AgarCyto specimens. A brush with cervical cells is suspended in primary fixative and transported (
Microscopic evaluation of AgarCytos was performed on a Leica Dialux transmission microscope (Leica; Wetzlar, Germany) equipped for digital image capture with a 3-CCD color video camera (DXC-950P; Sony, Tokyo, Japan) and a computer-interfaced frame grabber (Intellicam; Matrox Electronic Systems, Dorval, QC, Canada). Composite figures were produced using Adobe Photoshop 5.0.
Line Probe Assay-Polymerase Chain Reaction
LiPA-PCR for simultaneous detection of 25 HPV types was performed as described (Kleter et al. 1999) and applied to AgarCytos of differently fixed cell lines: HeLa, containing HPV 18, SiHa and CaSki, containing HPV 16, and A431, not containing any HPV type (1). The use of LiPA-PCR for Unifix-fixed cervical scrapings was recently validated (Melchers et al. 1999). Briefly, a single 4-± Agar-Cyto section was incubated overnight with proteinase K (0.3 mg/ml) and 10 μl of the sample was used for PCR. The PCR uses 5′-end biotinylated primers and produces 65-bp fragments from the HPV L1 open reading frame. The biotinylated PCR products were reverse-hybridized to a membrane strip containing 25 HPV specific oligonucleotide sequences equally spaced over the length of the strip. After hybridization and stringency washes, the biotin label was detected by alkaline phosphatase-conjugated streptavidin and visualized by a nitroblue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate (NBT/BCIP) reaction. HPV types were determined by comparing test samples with a positive control hybridization with all 25 HPV types present in the sample. Hybridization specificity was tested by hybridization with negative PCR controls.
DNA Image Cytometry (ICM)
DNA ICM requires a microscopic preparation technique that preserves nuclear integrity. Therefore, to include DNA image cytometry in a multiparameter analysis of a cervical scraping, a 200-μl sample has to be taken from the primary fixed cell suspension for cytospin preparation before Agar-Cyto embedding. We used Molt-4 and A431 cell lines for optimization of the sample preparation. The cell suspensions were primarily fixed for 16 hr in (a) 50% ethanol/2% carbowax, the standard cell fixative for DNA image cytometry (Bibbo 1991), (b) 70% ethanol, or (c) Unifix, and centrifuged at 2000 rpm for 5 min, using a Cyto-Tek cytocentrifuge (Sakura). Nuclear DNA was stoichiometrically stained as described (Schulte and Wittekind 1989). Briefly, cytospin preparations were postfixed in Böhm's fixative (85% methanol/4% formaldehyde/5% acetic acid) followed by rinsing in 70% methanol, 50% methanol, and distilled water. Cell preparations were consecutively hydrolyzed in 5 N HCl for 1 hr, rinsed in distilled water, stained with Feulgen-Schiff reagents for 1 hr, rinsed in running tap water for 20 min, dehydrated through ethanol series and xylol, and mounted in Permount. DNA index measurements were performed on a CAS 100 image analysis system (Becton Dickinson; Franklin Lakes, NJ). The experimental reference DNA index (DI) value for Molt-4 and A431 cell is given by the ethanol/carbowaxfixed cells.
Results
AgarCyto Cell Block Preparation
We optimized the cell block preparation for exfoliate cell material with respect to cell yield by (a) pelleting the cell suspension in agar in a 1.5-ml Eppendorf tube (Figures 1A-1C) as described by Kung et al. (1990) and (b) dividing the solid agar cone into two halves and embedding them side by side in paraffin (Figures 1D and 1E). This resulted in two compact, easy-to-find areas containing hundreds of cells for evaluation. One will typically use the X10 and X25 objectives to find the cell groups and cells of interest, making the screening time about 3 min per AgarCyto slide.
Cytomorphology
AgarCyto cell block sections of differently fixed cell lines with known morphological features were H&E-stained for morphological evaluation (1). The ethanol-containing fixatives (ethanol/carbowax, ethanol/carbowax/formaldehyde, and CytoRich) gave good cell morphology if the fixation lasted no longer than 16 hr. The cell shape was maintained and a normal nucleocytoplasmic ratio was observed. Longer fixation times compromised morphology. A fair morphology was observed after fixation in ethanol/carbowax or CytoRich for 16 hr or in ethanol/carbowax/formaldehyde for 64 hr. Neutral-buffered formaldehyde resulted in shrinkage of the cells and therefore overall poor morphology. In contrast, Unifix, a modified formaldehyde fixative, gave good morphology even when the cells were fixed for 64 hr. Cytomorphology of cervical scrapings after fixation in Unifix and AgarCyto processing was well preserved (see Figure 3A), and dysplastic groups were easily recognized. The morphological features used for the classification of Pap smears also apply to AgarCytos, with minor modifications. A comprehensive comparison between cytomorphology of Pap smears and AgarCytos is pending and will be published elsewhere.
Immunocytochemistry
The influence of different fixatives and fixation times on immunocytochemical staining for Ki-67 and p53 using standard laboratory protocols was monitored with regard to signal intensity, background staining, and staining efficiency Table 1. The cell morphology was not altered by the immunostaining procedure. Using the ethanol-based fixatives, Ki-67 immunodetection resulted in weak to moderate staining of the nuclei in only a small fraction of the cells, and the cells showed a high background staining. Prolonged fixation in ethanol-based fixatives generally decreased the intensity of immunostaining. Cells fixed in neutral-buffered formaldehyde gave strong, specific nuclear staining for Ki-67 in a representative fraction of the cells, but also showed some background. Best results were obtained with Unifix-fixed cells, which gave strong Ki-67 immunostaining without any background in a representative fraction of the cells.
P53 immunostaining of the positive control cell line A431 was strongly positive with all fixation protocols. Antigen retrieval steps were required to detect low levels of wild-type and mutant p53 in cervical tumor cell lines and Molt-4, respectively. Using different fixation protocols, we observed differences in the staining efficiency and signal-to-noise ratio for the detection of low-level p53. Immunostaining for p53 was weak in ethanol/carbowax and negative in CytoRich. In contrast, fixation with ethanol/carbowax/formaldehyde gave moderate p53 immunostaining, with a lower but still considerable background. Longer fixation times decreased the intensity of immunostaining. Neutral-buffered formaldehyde and Unifix gave clear, specific nuclear staining for p53 in a representative fraction of the cells. Best results were obtained with fixation in Unifix for 64 hr, because the signals were clearly observed without any background staining. Using the Unifix fixation protocol, AgarCyto preparations of cervical scrapings gave good signal-to-noise ratios for Ki-67 and p53 in dysplastic groups (Figure 3BC).
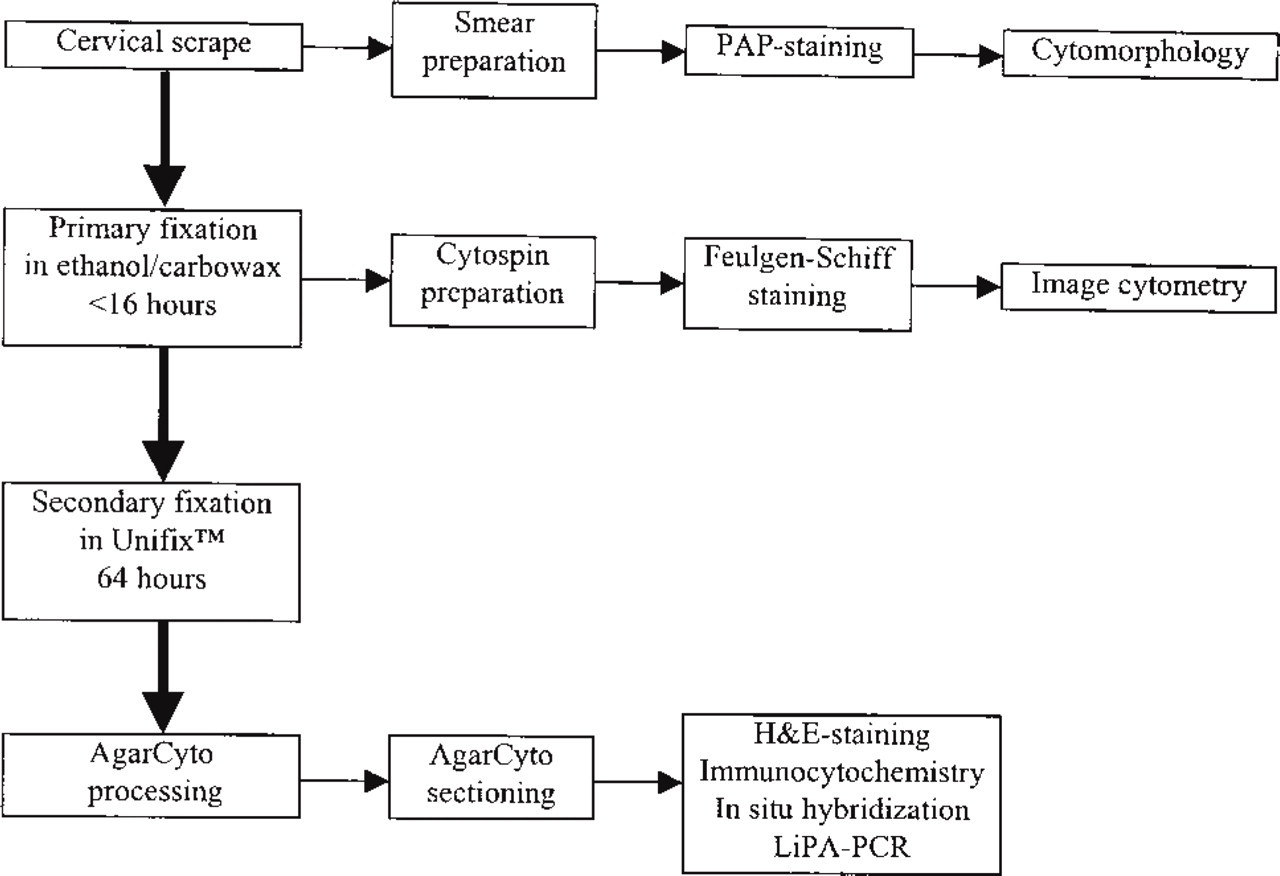
Flow chart of the AgarCyto cell processing method for multiple molecular techniques, also including Pap smear preparation for conventional cytomorphological screening and cytospin preparation for DNA image cytometry.
In Situ Hybridization
ISH results for the centromere of chromosome 1 (cen 1) and HPV Type 16 (HPV 16) were evaluated on the basis of signal intensity, signal shape, nuclear morphology, and number of ISH signals as known from the literature (1). To facilitate probe penetration, crosslinked proteins were removed by a proteolytic enzyme pretreatment. To standardize the experimental protocol, the differently fixed AgarCytos were all pretreated equally with sodium isothiocyanate and pepsin. Fixation for 2 hr in ethanol/carbowax or CytoRich resulted in moderate to strong ISH signals and good nuclear morphology. However, fixation for 16 or 64 hr resulted in fuzzy ISH signals that were difficult to discern. Ethanol/carbowax/formaldehyde fixation for 2 hr showed better results: discrete ISH signals and good nuclear morphology were observed, but the signal intensity decreased rapidly with prolonged fixation. Neutral-buffered formaldehyde resulted in strong and discrete ISH signals, and although overall cell morphology was poor (cell shrinkage), the nuclear morphology was fair. Best results were obtained with Unifix after 64 hr of fixation, because strong and discrete ISH signals were observed and overall cell morphology was very well maintained. Consistent ISH results for cen 1 and HPV 16 were obtained on AgarCyto sections of cervical scrapings that underwent the Unifix fixation protocol. In dysplastic cell groups, the ploidy status for cen 1 could be determined (Figure 3D). HPV 16 ISH combined with CARD signal amplification specifically detects HPV 16 sequences in individual cells and is able to discriminate episomal from integrated infection (Figure 4).
LiPA-PCR
DNA isolated from a single 4-±-thick AgarCyto section has proved sufficient for reliable HPV screening with LiPA-PCR, irrespective of the type of fixative (1). A431 that does not contain any HPV sequences was always negative. Examples of HPV detection and typing by LiPA-PCR in AgarCyto cervical scrapings are given in Figure 5.
DNA Image Cytometry
To allow DNA ICM of whole nonsectioned cells, a small fraction of the primarily-fixed cell suspension was reserved for cytocentrifugation before further processing into AgarCytos (Figure 2). We compared two fixatives (70% ethanol and Unifix) with the standard fixative (50% ethanol/2% carbowax) concerning the DNA index (DI) on Molt-4 and A431 cell lines (Table 2). After ethanol/carbowax fixation, the DI of Molt-4 cells was 2.17 and of A431 cells was 2.02. The DI of Molt-4 was 2.16 after 70% ethanol fixation and 2.00 after Unifix fixation, which is lower than the reference value. For A431 cells, the DI values differed from their reference DI value more prominently, i.e., 1.92 for 70% ethanol and 1.86 for Unifix. Moreover, Unifixfixed cells did not stick well to the glass slides. Ethanol/carbowax fixation gave the best results for DNA ICM. For further processing of the remaining cells into AgarCytos, a secondary fixation in Unifix is required within 16 hr to maintain good cytomorphology and successful application of ICC and ISH.
Discussion
We have developed the AgarCyto cell processing method for multiple diagnostic analyses of a single brush sample from the uterine cervix. The AgarCyto processing method includes preparation of a routine Pap smear and the collection and processing of the remaining cells on the brush in paraffin. Multiple microscopic specimens can be produced from a single scraping for the application of standard H&E staining, ICC, and ISH. Furthermore, a single AgarCyto section can serve as DNA input for LiPA-PCR and, before AgarCyto embedding, cytospin preparations can be produced for DNA image cytometry.
In diagnostic cytology, it has been advocated that molecular techniques will improve cytopathological diagnosis and may predict clinical course. Up to now, diagnostic molecular markers for cervical cancer have been identified from retrospective studies on histological material or re-stained archival Pap smears. Prospective studies on cytological material are hampered by insufficient amounts of diagnostic specimens from the same patient. Cell block processing methods of cytological material may solve this problem, but none of the developed methods describes comprehensive application of molecular techniques in a routine setting.
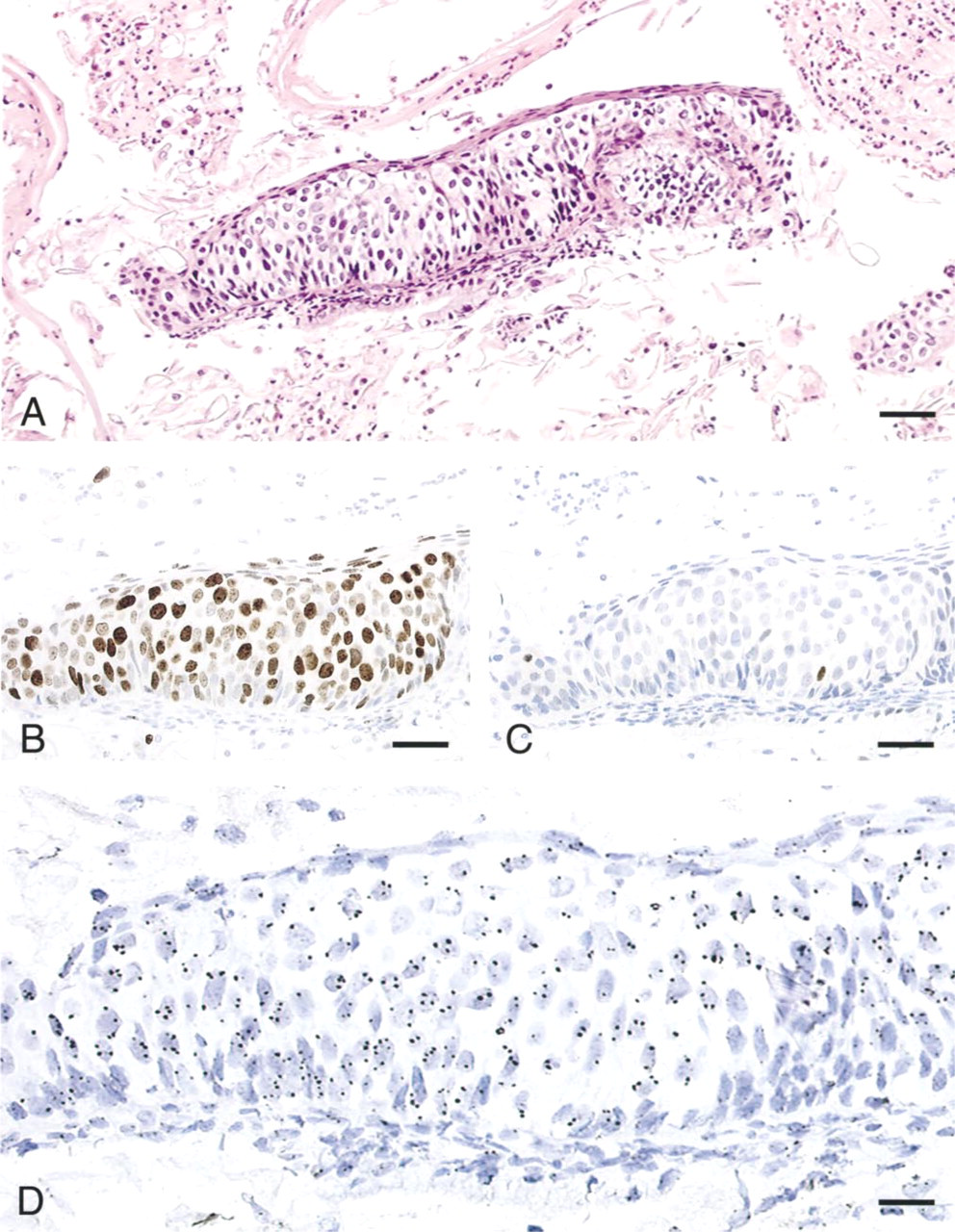
Serial sections of an AgarCyto of a cervical scraping diagnosed as Pap 4 in conventional cytology. The cells were fixed in Unifix and subjected to standard staining protocols. The H&E staining shows excellent cell morphology. The dysplastic cell group shows disturbed nucleocytoplasmic ratios and irregular chromatin (
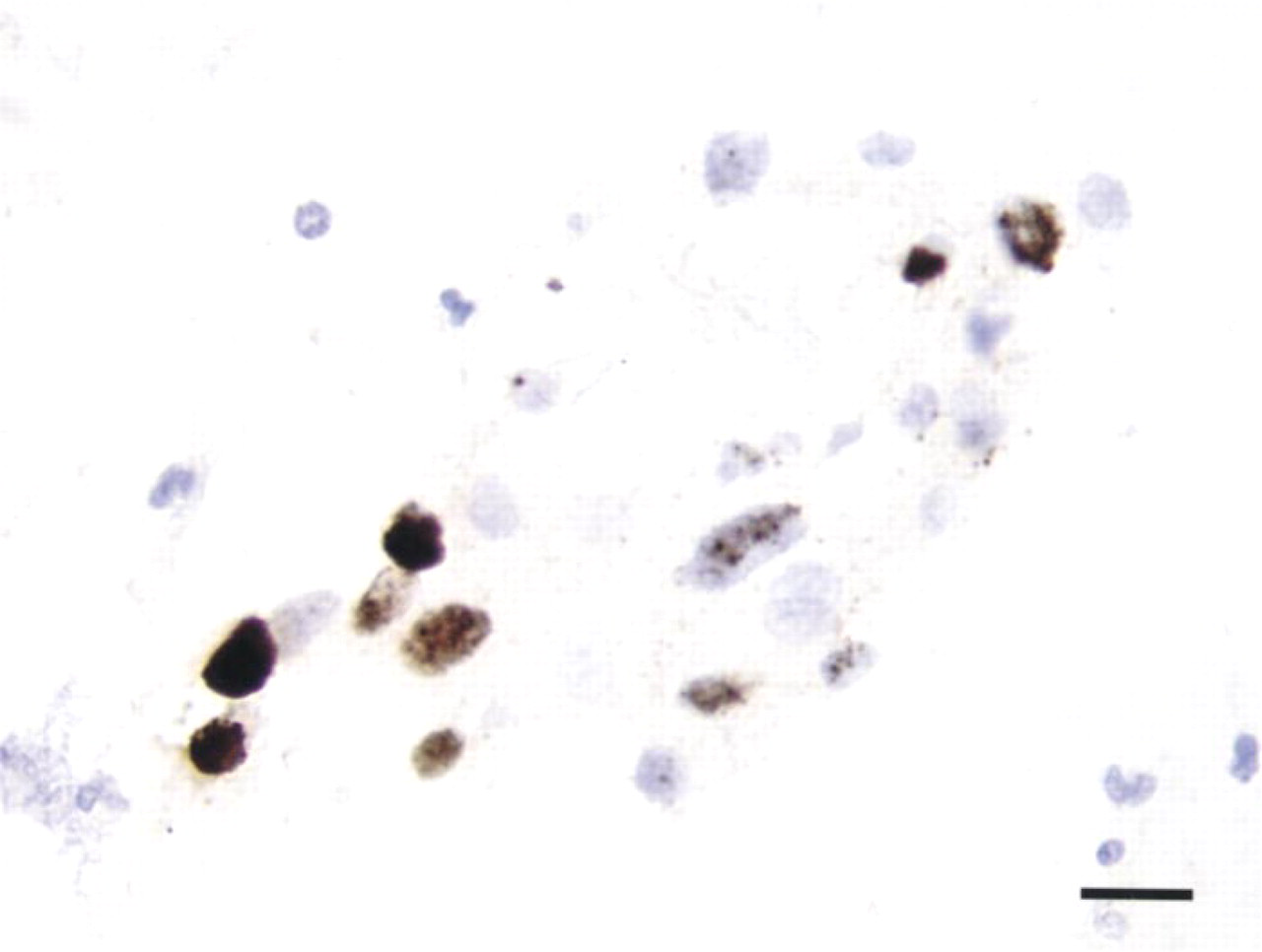
ISH on an AgarCyto of a cervical scraping for HPV 16 using CARD detection shows a nuclear staining pattern characteristic of integrated HPV sequences. Bar = 20 ±.
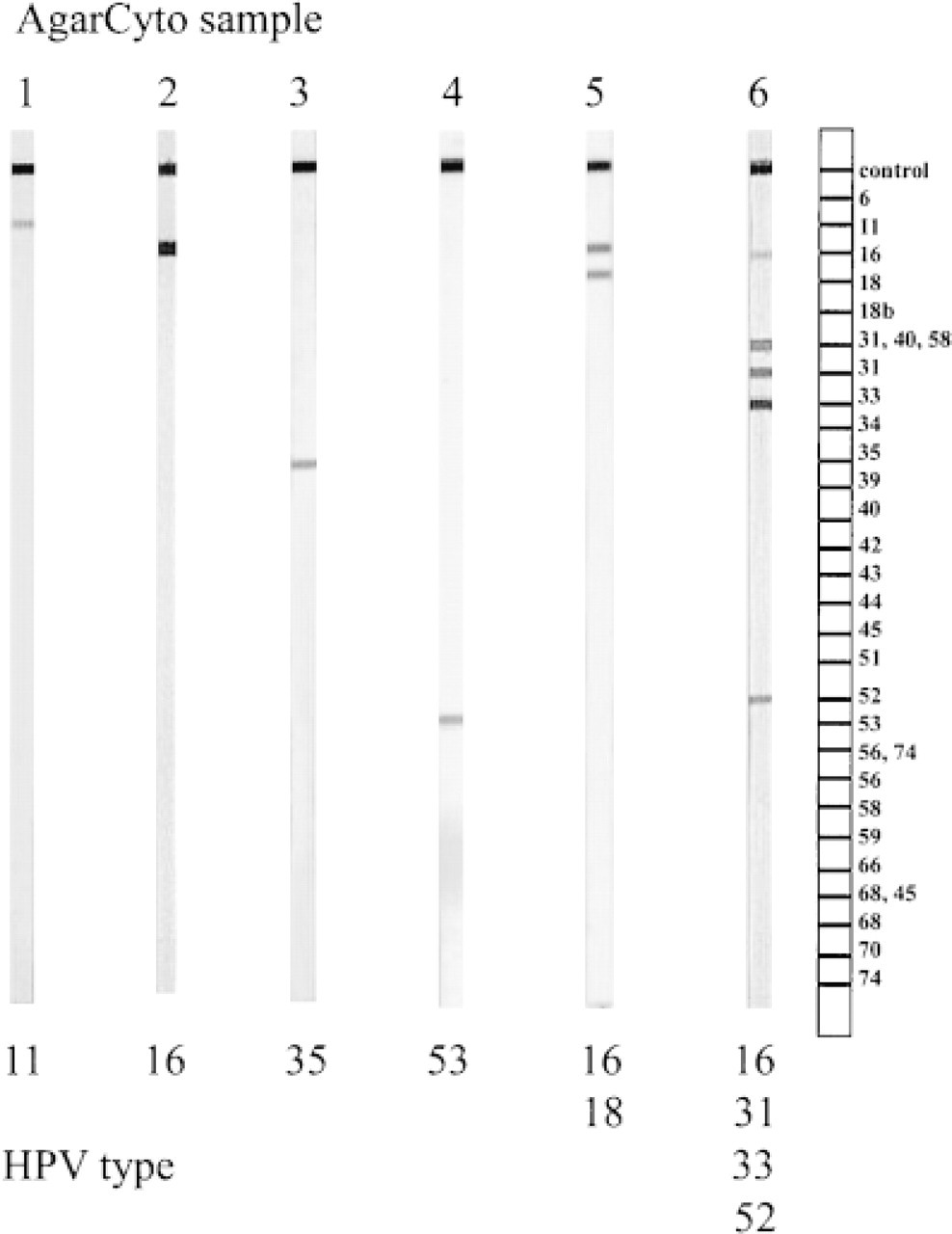
Summary of LiPA-PCR typing of HPV for six selected patient samples with known HPV infection status. Cervical scrapings were fixed in Unifix and processed into AgarCytos. One 4-± section was sufficient for unambiguous HPV detection and typing.
Our methodological research has focused on the application of a wide range of molecular diagnostic techniques, using both in situ techniques, while maintaining cell morphology, and biochemical techniques.
The protocol for AgarCyto processing of cervical scraping material is summarized in Figure 2. We recommend primary fixation of the cells in ethanol/carbowax. The cell suspension can be transported and, if so desired, cytospin preparations can be produced for DNA image cytometry. Cells must be secondarily fixed in Unifix within 16 hr, however, to ensure good cell morphology in ICC and ISH staining of the AgarCyto. Therefore, if DNA image cytometry is not performed, primary fixation in Unifix is preferred.
The fixative that yielded the best overall results was Unifix, a 4% formaldehyde-based fixative containing zinc sulfate, methanol, and other constituents. We used standard protocols for ICC and ISH with each fixation type. It is conceivable, however, that by adjusting the ICC and ISH protocols optimal results would have been obtained with each fixation. In particular, ethanol/carbowax and ethanol/carbowax/formaldehyde fixation for 2 hr yielded good overall results that may need slight modification of the ICC protocol. However, short fixation times are not preferred in routine laboratory practice. Cervical scrapings are usually obtained in the clinic and must be transported to the diagnostic laboratory. Therefore, 16-hr (i.e., overnight) ethanol/carbowax and subsequent Unifix fixation, which is allowed to last 64 hr (i.e., over the weekend), provides ideal logistics and yields the best overall results.
Influence of fixation on the DNA index measurements of Molt-4 and A431 cells a
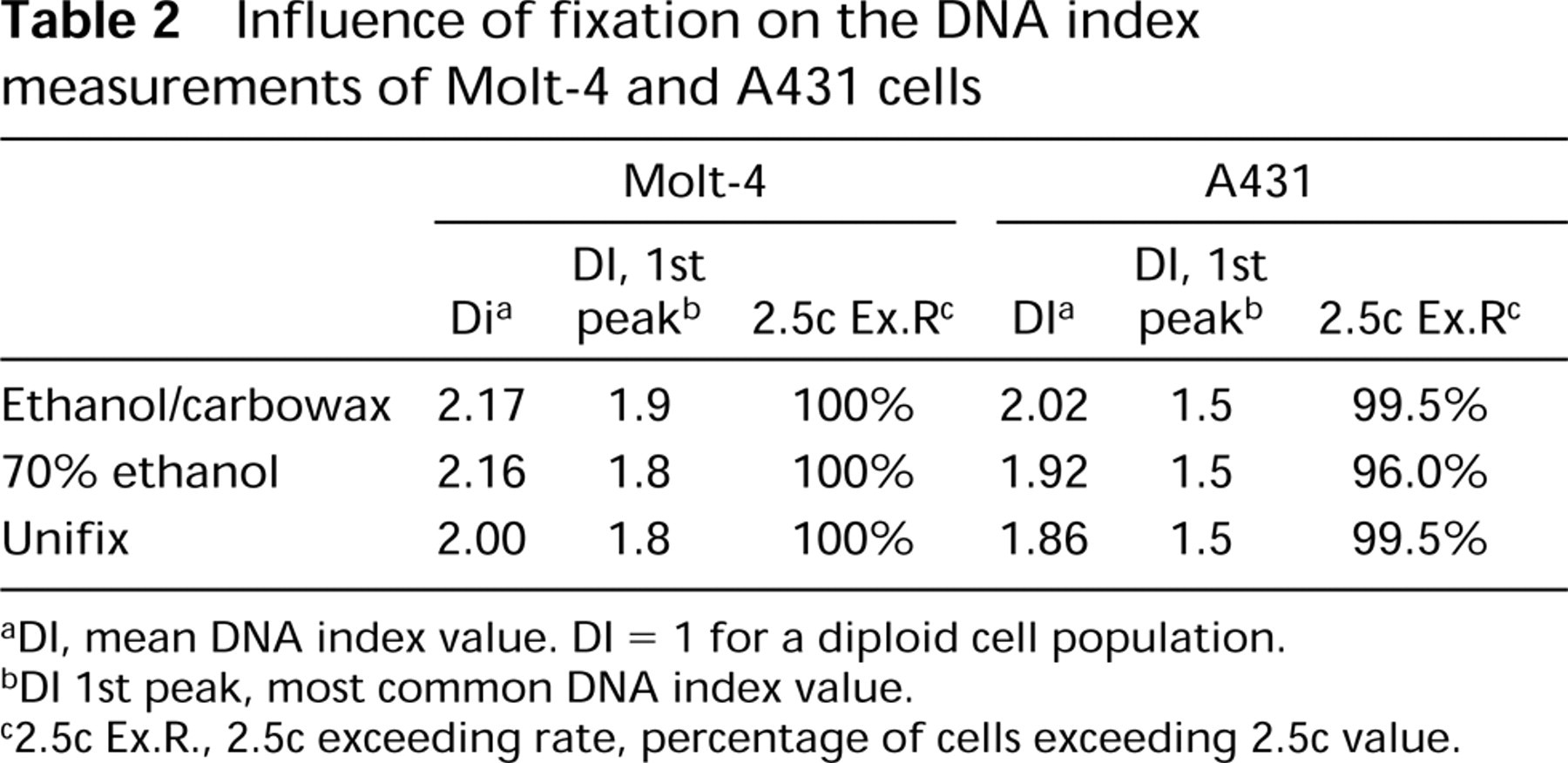
aDI, mean DNA index value. DI = 1 for a diploid cell population.
bDI 1st peak, most common DNA index value.
c2.5c Ex.R., 2.5c exceeding rate, percentage of cells exceeding 2.5c value.
Sensitivity of the immunostaining procedure for p53 was dependent on the use of fixation and antigen retrieval methods (Fisher et al. 1994). Without antigen retrieval, mutant p53 could be detected only in abundantly expressing A431 cells but not in the other cell lines (not shown). In contrast, with antigen retrieval, immunodetection was feasible of low-level wild-type p53 in HPV-containing cervical cell lines and low-level mutant p53 in Molt-4 cells. Using this standardized procedure for cervical scrapings, we are able to detect low levels of both mutant and wild-type p53.
HPV typing of the cervical cell lines by LiPA-PCR was highly sensitive and specific, regardless of the type of fixation. This result underlines the robustness of the LiPA-PCR technique. The application of LiPA-PCR, using input DNA from AgarCytos of a large group of women diagnosed with a cervical neoplasia, will be discussed elsewhere (Melchers et al. 1999).
Our findings with Unifix fixation are in accordance with earlier reports on the use of Zn-containing formaldehyde fixatives. Good results have been obtained with ISH for HPV 16 DNA in CaSki and SiHa cells (Tbakhi et al. 1998) and with PCR establishing B-cell clonality (Tbakhi et al. 1999). The use of Zn-formaldehyde fixations has been found to give excellent results in immunostaining of histological samples in many laboratories around the world (Dapson 1993) and has now proved its value for cytological material.
The AgarCytos could be valuable for routine cytomorphological diagnosis. Although the preparation procedure is more time-consuming than smears or monolayers and the cytomorphology of AgarCytos looks somewhat different from routine smear preparations due to sectioning, a favorable feature of AgarCyto morphology is the histology-like appearance (Figure 3A). Cell clumping, as often observed in smears and causing dubious diagnoses, are easier to interpret in AgarCyto sections because cell polarity and differentiation can be examined just as in histology. AgarCyto diagnosis may be of use if smear preparations are inadequate because of too many cell clumps. However, clinical studies need to be performed to validate cytopathological diagnosis based on AgarCytos. Once the diagnostic cytomorphological criteria of AgarCytos have been determined, AgarCytos will be the ideal vehicle for molecular inventory studies. Both retrospective and prospective studies can be employed to identify biomarkers of premalignant lesions of cervical cancer and other types of cancer that depend on screening of cytological material. In our experience, AgarCyto cell processing can be applied equally well to uterine cervix brushes, oral cavity brushes, urine, bladder washes, lung sputa, bronchial washes, and fine-needle aspiration samples. Therefore, AgarCytos cell processing should become a routine cytological laboratory practice.
Footnotes
Acknowledgements
Acknowledgments
Supported by the Dutch Cancer Society KUN 97-1486.
We wish to thank A. Gemmink and S. Wienk for excellent technical assistance, I. Cornelissen for culturing cells, H. De Leeuw for DNA image cytometry measurements, and Prof Dr J. Merkus, Dr B. Keijzer, Dr L. Massuger, and C. Schijf for valuable scientific discussions.