
Abstract
PtdIns(4,5)P2 and PtdIns(4,5)P2 5-phosphatases play important roles in diverse aspects of cell metabolism, including protein trafficking. However, the relative importance of the PtdIns(4,5)P2 5-phosphatases in regulating PtdIns(4,5)P2 levels for specific cell processes is not well understood. Ocrl1 is a PtdIns(4,5)P2 5-phosphatase that is deficient in the oculocerebrorenal syndrome of Lowe, a disorder characterized by defects in kidney and lens epithelial cells and mental retardation. Ocrl1 was originally localized to the Golgi in fibroblasts, but a subsequent report suggested a lysosomal localization in a kidney epithelial cell line. In this study we defined the localization of ocrl1 in fibroblasts and in two kidney epithelial cell lines by three methods: immunofluorescence, subcellular fractionation, and a dynamic perturbation assay with brefeldin A. We found that ocrl1 was a Golgi-localized protein in all three cell types and further identified it as a protein of the trans-Golgi network (TGN). The TGN is a major sorting site and has the specialized function in epithelial cells of directing proteins to the apical or basolateral domains. The epithelial cell phenotype in Lowe syndrome and the localization of ocrl1 to the TGN imply that this PtdIns(4,5)P2 5-phosphatase plays a role in trafficking.
Keywords
A
Recently, attention has focused on the role of PtdIns (4,5)P2 phosphatases in vesicle trafficking (Liscovitch and Cantley 1995; DeCamilli et al. 1996; Fang et al. 1998). Synaptojanin is one member of this protein family that appears to participate in membrane trafficking at synaptic vesicles (McPherson et al. 1995; Chung et al. 1997). Another member of this family, ocrl1, was localized to the Golgi complex (Olivos-Glander et al. 1995). Since each of the Golgi compartments, cis, medial, trans, cis-Golgi network (CGN), and trans-Golgi network (TGN), plays a different role in sorting, the localization of this PtdIns(4,5)P2 5-phosphatase within the Golgi complex is important in uncovering its function. This study used a well-established metabolic perturbation assay combined with immunofluorescence and subcellular fractionation of several cell lines to determine that ocrl1 is localized specifically to the TGN region of the Golgi complex.
Materials and Methods
Cell Culture
Normal human fibroblast cell lines (CRL1489) and Vero monkey kidney epithelial cells (CCL81) were obtained from American Type Culture Collection (Manassas, VA). Human fibroblasts, Vero cells, and mouse fibroblasts derived in our laboratory (Jänne et al. 1998) were grown in Dulbecco's modified essential medium (GIBCO; Grand Island, NY) with 10% fetal bovine serum and 2 mM glutamine at 37C with 5% CO2. A normal human kidney epithelial cell line (NHK52) bearing a temperature-sensitive SV40 large T-antigen plasmid was provided by Dr. L.C. Racusen (Racusen et al. 1995). NHK cells were maintained in media containing 50% Ham's F12, 50% Dulbecco's modified Eagles' medium, 5% fetal bovine serum, 25 mM Hepes, 2.5 μg/ml insulin, 50 ng/ml hydrocortisone, 5 μg/ml transferrin, 5 ng/ml sodium selenite, 20 ng/ml triiodothyronine, 50 U/ml penicillin, 50 μg/ml streptomycin, and 2.5 μg/ml amphotericin. The cells were maintained at 33C in a humidified atmosphere of 95% air, 5% CO2 until plating for studies, at which time they were shifted to 37C.
Antibodies
The antibodies used for immunofluorescence studies were anti-α-tubulin, anti-γ-adaptin (Sigma Chemical; St Louis, MO), anti-lamp1 (clone H4A3) (Developmental Studies Hybridoma Bank; University of Iowa, Ames, IA), and an affinity-purified polyclonal anti-ocrl1 antibody (Olivos-Glander et al. 1995). For confirmation of subcellular fractions by Western blotting, antibodies to the following organelle-specific proteins were used: the nuclear vitamin D receptor (Affinity Bioreagents; Golden, CO); mitochondrion-specific cytochrome c oxidase (Molecular Probes; Eugene OR), and hsp 70 (Affinity Bioreagents); the lysosomal marker lamp1; Golgi-localized golgin-84 (Bascom et al. 1999) and TCP-1 (Affinity Bioreagents); the endoplasmic reticulum marker protein disulfide isomerase (Affinity Bioreagents); and two vesicle-associated proteins, β-COP (kindly provided by Dr. J. Lippincott-Schwartz, NICHD, NIH) and clathrin (Maine Biotechnology Services; Portland, ME). The mannose 6-phosphate receptor antibody was a kind gift from Dr. J. Paul Luzio (Cambridge University; Cambridge, UK).
Immunofluorescence microscopy
Fibroblasts and Vero cells were grown on LabTek chamber slides (Naperville, IL) coated with 0.01% poly-
NHK cells were plated on the coated chamber slides and switched from 33C to 37C for 3-5 days to allow differentiation before immunocytochemistry. Immunocytochemistry was performed at room temperature. Cells were fixed and permeabilized with 1% paraformaldehyde, 0.1% Triton X-100 in PBS for 5 min, washed with PBS, and blocked with 5% goat serum (Gibco/BRL; Gaithersburg, MD) for 30 min. The cells were incubated with primary antibody for 1.5 hr washed 3 times with PBS, and incubated with secondary antibody, Alexa 488, or Alexa 594 (Molecular Probes) for 30 min. Fluorescence was visualized on a Leica DMR fluorescence microscope.
Sucrosome Formation and Vital Staining of Cell Lysosomes
Cells were incubated in 100 mM sucrose in growth medium for 1-3 days and washed with PBS (Karageorgos et al. 1997). For vital staining of lysosomes, cells were incubated with LysoTracker™ (Molecular Probes) for 45 min, washed with PBS, and fixed in 1% paraformaldehyde. LysoTracker™ is a vital dye that stains lysosomes on the basis of their acidic pH.
Subcellular Fractionation
Human kidney (NHK) and mouse fibroblasts were homogenized with a ball-bearing homogenizer in 0.25 M sucrose, 10 mM Tris-HCl (pH 7.4) with 4 μg/ml aprotinin, 4 μg/ml leupeptin, and 100 mg/ml phenylmethylsulfonylfluoride (Balch et al. 1988). Subcellular fractionation was performed by differential centrifugation based on a method previously described (Koenig 1974). The homogenate was centrifuged at 600 times; g for 10 min and the pellet containing the plasma membrane and nuclei was saved. The remaining supernatant was centrifuged at 12,000 times; g for 20 min. and the pellet saved as the mitochondrial/lysosomal fraction. The resulting supernatant was then centrifuged at 100,000 times; g for 1.5 hr to obtain the Golgi/microsome pellet and the cytosolic (supernatant) fractions.
The fractions were tested to confirm their enrichment for specific organelles by Western analysis (Olivos-Glander et al. 1995) with organelle-specific antibodies.
Golgi Perturbation Studies
For the Golgi perturbation studies, fibroblasts were incubated with 5 μg/ml brefeldin A (BFA) (Epicentre Technologies; Madison, WI) for 1 hr at 37C in a humidified atmosphere of 95% air, 5% CO2, as reported previously (Lippincott-Schwartz et al. 1991). Vero and NHK cells were incubated with 15 μg/ml BFA. Cells were fixed, permeabilized, and immunocytochemistry was performed.
Results
Immunofluorescence
To compare the localization of ocrl1 in fibroblasts and kidney epithelial cells, three cell lines were studied by immunofluorescence with the ocrl1 antibody, an antibody to the TGN protein γ-adaptin, and two lysosomal markers, lamp1 antibody and LysoTracker™. Figure 1 shows double-label immunofluorescence of ocrl1 (green) and γ-adaptin staining (red) of normal human fibroblasts (Figures 1A-1C) and of kidney epithelial cells of monkey (Vero cells) (Figures 1D-1F) and human origin (NHK cells) (Figures 1G-1I). In human fibroblasts, ocrl1 staining (Figure 1A) was distributed in a juxtanuclear reticular pattern typical of a Golgi complex protein, very similar to that of γ-adaptin staining (Figure 1B). In fact, there appeared to be significant co-localization (yellow staining) of ocrl1 and γ-adaptin to the juxtanuclear region of fibroblasts and kidney epithelial cells, as shown in the double exposures (Figures 1C, 1F, and 1I). The overlap of ocrl1 with γ-adaptin was more striking than that with β-COP reported previously (Olivos-Glander et al. 1995).
In addition to the predominantly juxtanuclear ocrl1 staining, there was a small amount of punctate staining in the cytoplasm of fibroblasts and Vero cells. This staining is specific because it was not observed in fibroblasts from Lowe syndrome patients, which lack ocrl1 (not shown). Punctate cytoplasmic staining was also observed with γ-adaptin (Figure 1) and clathrin immunofluorescence (data not shown), but the cytoplasmic γ-adaptin and clathrin staining were not coincident with the cytoplasmic ocrl1 staining.
Because a previous report proposed a lysosomal localization of ocrl1 in NHK cells (Zhang et al. 1998), we performed double-label immunofluorescence with ocrl1 and two lysosomal markers, lamp1 and LysoTracker™. To help distinguish lysosomes from other organelles, cells were grown in sucrose before staining for lysosomes. Because mammalian cells do not metabolize sucrose, sucrose incubation results in swollen lysosomes (Karageorgos et al. 1997; Dinter and Berger 1998). Figure 2 shows ocrl1 (green) and lamp1 staining (red) in fibroblasts (Figures 2A-2C) and Vero cells (Figure 2D-2F). The lamp1 staining in fibroblasts (Figure 2B) and Vero cells (Figure 2E) is a patchy cytoplasmic staining and is not concentrated in the perinuclear region as is the ocrl1 staining. The double exposures emphasize the fact that the ocrl1 staining is perinuclear and quite different from the patchy cytoplasmic staining of lamp1 (Figures 2C and 2F).
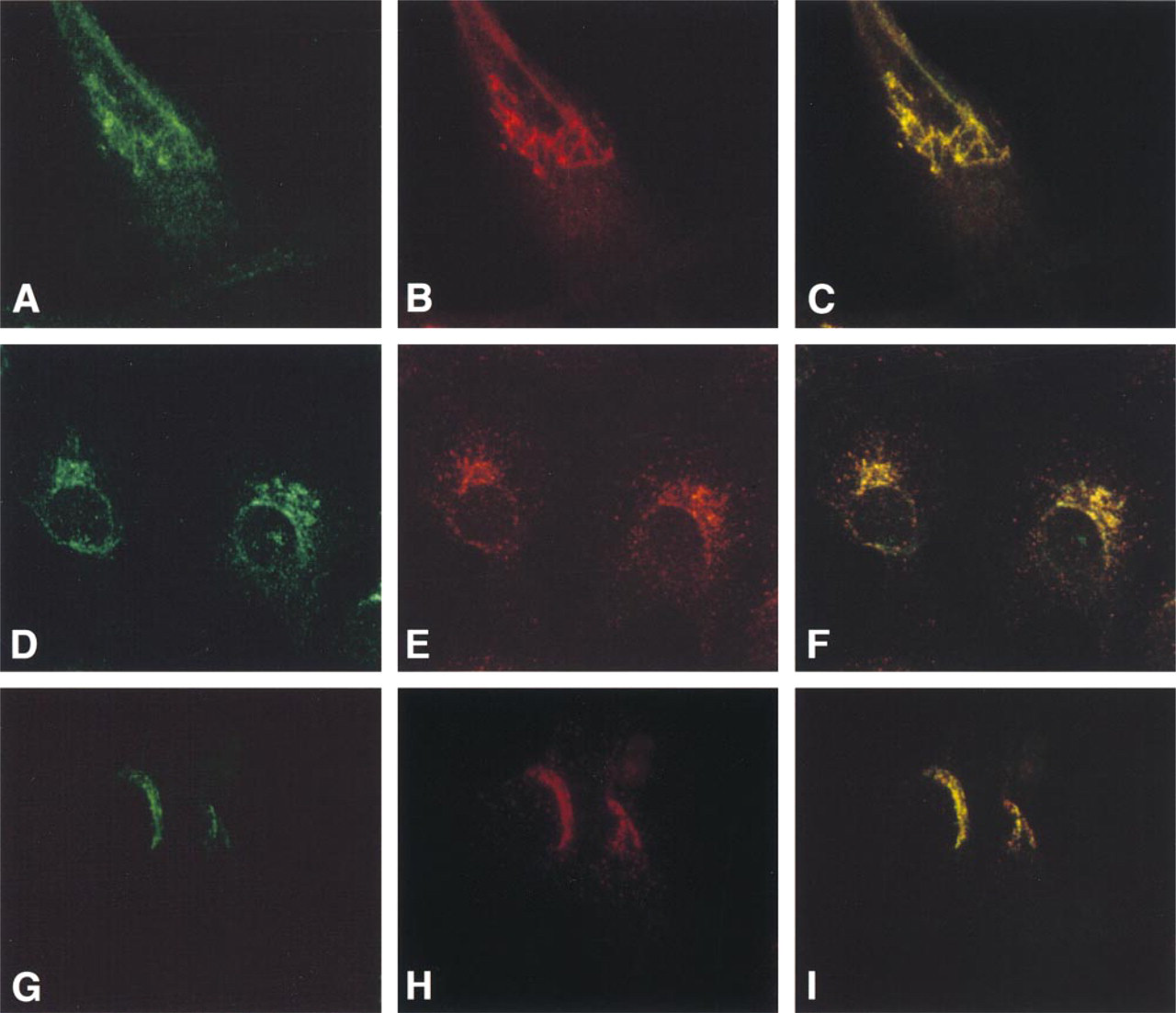
Immunofluorescence co-localization of ocrl1 and γ-adaptin to the Golgi complex in double-labeled fibroblasts (
To confirm that ocrl1 was not localized to lysosomes in fibroblasts and Vero cells, we performed immunocytochemistry with a second lysosomal marker, the vital lysosomal dye LysoTracker™. As with the lamp1 staining, the LysoTracker™ staining was cytoplasmic, with a patchy distribution in both fibroblasts (Figure 2H) and Vero cells (Figure 2K). The double exposures (Figure 2I and 2K) show a distinct separation between the two staining patterns, with ocrl1 exhibiting a more polarized staining pattern than the LysoTracker™ staining. These immunofluorescence studies clearly demonstrate that ocrl1 is not localized to lysosomes in these cells.
We then compared the staining of ocrl1 and lamp1 in NHK cells, the cells used in a previous report to localize ocrl1 to lysosomes (Zhang et al. 1998). In NHK cells, ocrl1 staining (green) was clustered near the nucleus as it was in the other cell types (Figure 2M). The lamp1 staining (red) in the NHK cells, however, had a different appearance from the patchy cytoplasmic staining seen in fibroblasts and Vero cells (Figures 2B and 2E). It was polarized and clustered near the nucleus (Figure 2N). The lamp1 staining appeared to partially intersect with ocrl1 staining in NHK cells (yellow staining), although there were also regions of ocrl1 and lamp1 staining that did not overlap (Figure 2O). Even though there was considerable overlap of ocrl1 and lamp1 staining, there was also a striking overlap between ocrl1 and γ-adaptin in these cells (Figure 1I). This made it clear that immunofluorescence studies alone were inadequate to determine the subcellular localization of ocrl1 in NHK cells. We therefore performed subcellular fractionation and Golgi perturbation experiments to determine the localization of ocrl1 in these cells.
Subcellular Fractionation
Subcellular fractionation was performed with the NHK human kidney cells and with mouse fibroblasts. The latter were used because of their rapid growth rate to test consistency of the fractionation procedure. Fractionation was performed to isolate nuclear, mitochondrial/lysosomal, Golgi/microsomal and cytosolic fractions. For confirmation of the fractions, Western analysis was performed with antibodies to proteins with a well-established subcellular localization. Western analysis confirmed that nuclei, mitochondria, and Golgi were consistently found in the expected fractions based on their separation by differential centrifugation (Koenig 1974) in four separate experiments in mouse fibroblasts.
The NHK cell fractionation results were similar to those of the mouse fibroblasts and are shown in Figure 3. Lamp1 and the mitochondrial marker hsp-70 were highly enriched in the mitochondrial/lysosomal fraction (Figure 3d). A small amount of lamp1, estimated at about 10%, was also present in the Golgi/microsomal fraction (Figure 3e). The integral Golgi membrane protein golgin-84 and the TGN protein γ-adaptin were enriched in the Golgi/microsomal fraction (Figure 3b and 3c). These proteins were also present in the mitochondrial/lysosomal fraction. The fact that the TGN and Golgi markers were in the mitochondrial/lysosomal fraction indicates that there may be some contamination of this fraction with Golgi or TGN elements. The Golgi/microsomal and mitochondrial/lysosomal fractions also contained the mannose 6-phosphate receptor (M6PR), indicating that endosomes were present in these fractions as well (Figure 3f). M6PRs are not found on lysosomes but are found on prelysosomes, late and early endosomes, and the TGN (Geuze et al. 1988; Griffiths et al. 1988, 1990). The localization of endosomes in these fractions would be expected on the basis of previous centrifugation studies (Griffiths and Simons 1986; Griffiths et al. 1988, 1990; Mullock et al. 1994; Akasaki et al. 1995; Tjelle et al. 1996). Two vesicle-associated proteins, clathrin and β-COP, were enriched primarily in the cytosolic fraction, representing isolated vesicles and/or soluble protein not associated with membranes (Figures 3g and 3h). Ocrl1 was enriched in the Golgi/microsomal fraction (Figure 3a). There was a striking similarily between the distribution of ocrl1 and the known TGN protein γ-adaptin in the fractions (Figure 3c). These subcellular fractionation data clearly demonstrated that ocrl1 and lamp1 were in different sub-cellular compartments in NHK cells and indicated that, as in other cell types, ocrl1 is a Golgi-associated protein.
Golgi Perturbation Experiments
We performed dynamic Golgi perturbation experiments with two purposes in mind. First, we wanted to provide independent evidence to confirm in NHK cells the immunofluorescence and subcellular fractionation data indicating that ocrl1 was a Golgi-associated protein. Second, we sought to identify the localization of ocrl1 within the Golgi apparatus. The fungal metabolite BFA disrupts the Golgi and redistributes components from Golgi stacks to the endoplasmic reticulum, while the TGN and early endosomes redistribute to the microtubule organizing center (MTOC) (Lippincott-Schwartz et al. 1991; Wood et al. 1991; Reaves and Banting 1992). Incubation of fibroblasts (Figures 4D-4F) and Vero cells (Figures 4J-4L) with BFA results in the redistribution of both ocrl1 (green) and the γ-adaptin (red) to a concentrated spot near the nucleus. Photographs of untreated fibroblasts (Figures 4A-4C) and Vero cells (Figures 4G-4I) from the same experiment are provided for comparison. The computer overlays show that the ocrl1 and γ-adaptin staining condense to the same spot (Figures 4F and 4L). To demonstrate that the intense spot of perinuclear staining corresponds to the MTOC, we double-labeled BFA-treated fibroblasts with ocrl1 (green) (Figure 4M) and the microtubule protein α-tubulin (red) (Figure 4N). Microtubules converge at the MTOC, and immunofluorescence staining with α-tubulin results in a concentrated juxtanuclear spot of staining. A computer overlay demonstrates that the spot of intense ocrl1 staining near the nucleus corresponds to the MTOC (Figure 4O, yellow staining).
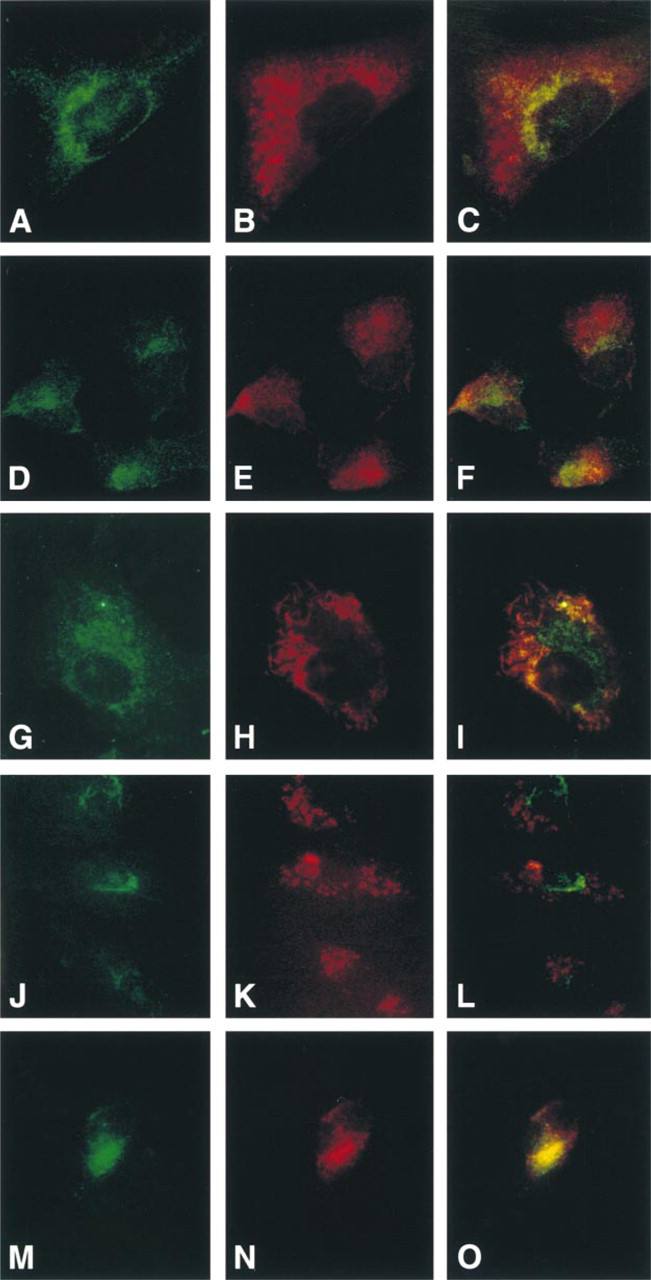
Immunofluorescence of ocrl1 and lysosomal markers in fibroblasts, Vero cells and NHK cells. Double-labeling of fibroblasts (
Since a previous study has demonstrated that lysosomal proteins do not redistribute to the MTOC with BFA treatment (Lippincott-Schwartz et al. 1991), we used this perturbation assay to confirm that lamp1 and ocrl1 were in different compartments in NHK cells. NHK cells were incubated with BFA and double-stained with ocrl1 (green) and lamp1 (red) antibodies. In most cells, the ocrl1 staining was concentrated in a spot near the nucleus (Figure 5A, solid arrowheads). However, the lamp1 staining retained its perinuclear clustered distribution, and did not condense to the MTOC (Figure 5B). Figure 5A includes a cell in which the ocrl1 staining appears dispersed and has not condensed to the MTOC (open arrowhead); this is likely to be a cell undergoing mitosis (Willison et al. 1989). The redistribution of ocrl1 to the MTOC after BFA incubation and the failure of lamp1 to redistribute in perturbation experiments confirms that these two proteins are in different subcellular compartments and demonstrates that ocrl1 is a TGN protein.
Discussion
There is ample evidence that PtdIns(4,5)P2 is an activator or cofactor for a number of membrane-trafficking events at the plasma membrane and in the Golgi complex (Terui et al. 1994; DeCamilli et al. 1996; Chung et al. 1997; Martin 1998; Erneaux et al. 1999). One of its main metabolic fates is dephosphorylation by one of a number of identified PtdIns(4,5)P2 5-phosphatases. The only PtdIns(4,5)P2 5-phosphatase that has been localized to the Golgi complex is the 105-kD 5-phosphatase ocrl1 (Olivos-Glander et al. 1995). This protein is deficient in the inherited disorder Lowe syndrome, resulting in defects in renal tubule cell function and lens development as well as mental retardation. It is possible that the restriction of the phenotype to certain polarized cells, such as lens and renal epithelial cells, results from defects in polarized epithelial cell function. The original studies localizing ocrl1 to the Golgi in fibroblasts (Olivos-Glander et al. 1995) were of interest because the Golgi plays a key role in sorting proteins to apical or basolateral membranes of polarized cells. However, a more recent report, also using immunocytochemical methods, localized ocrl1 to lysosomes in a human kidney proximal tubule cell line (Zhang et al. 1998). In the present study we sought to confirm the Golgi localization in fibroblasts by several methods, to assess its localization in kidney epithelial cells by using more rigorous methods than those used previously, and to extend our original observations to determine the Golgi compartment to which this PtdIns(4,5)P2 phosphatase is localized. Attempts at immunoelectron microscopy with the ocrl1 antibody were inadequate. The ocrl1 antibody, made against the denatured protein, was used with mild fixation conditions for the immunofluorescence experiments but reacted poorly with tissue that had been sufficiently fixed and permeabilized to allow the preservation of morphology for electron microscopy. In the immunofluorescence experiments shown here, a much lower dilution and longer exposure times were used for the ocrl1 antibody compared with the γ-adaptin antibody.
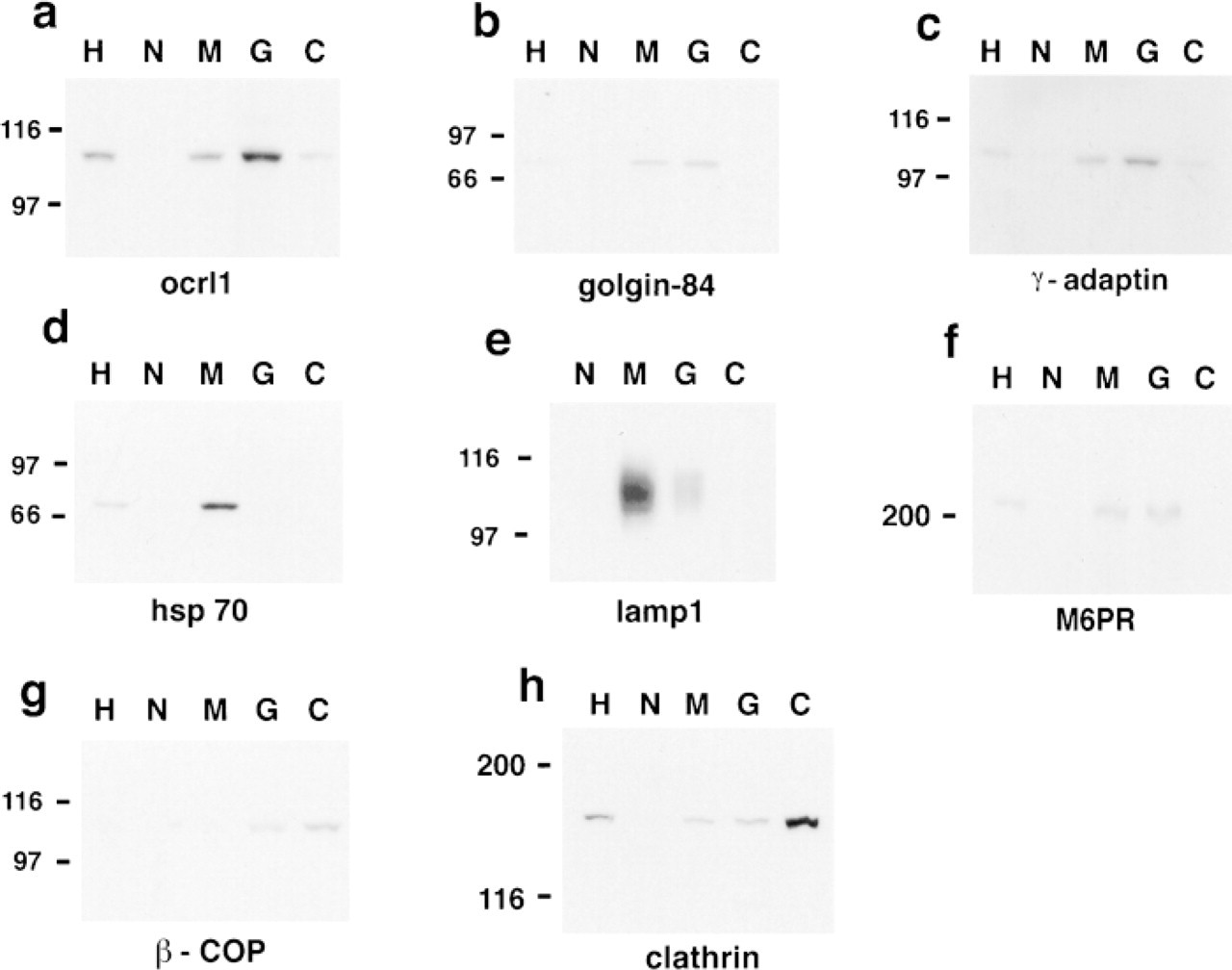
Subcellular fractionation of NHK cells. Fractions were obtained by differential centrifugation in 0.25 M sucrose, 10 mM Tris, pH 7.4, as described in the text and were analyzed by Western blotting. Equal amounts of protein were loaded in each lane:
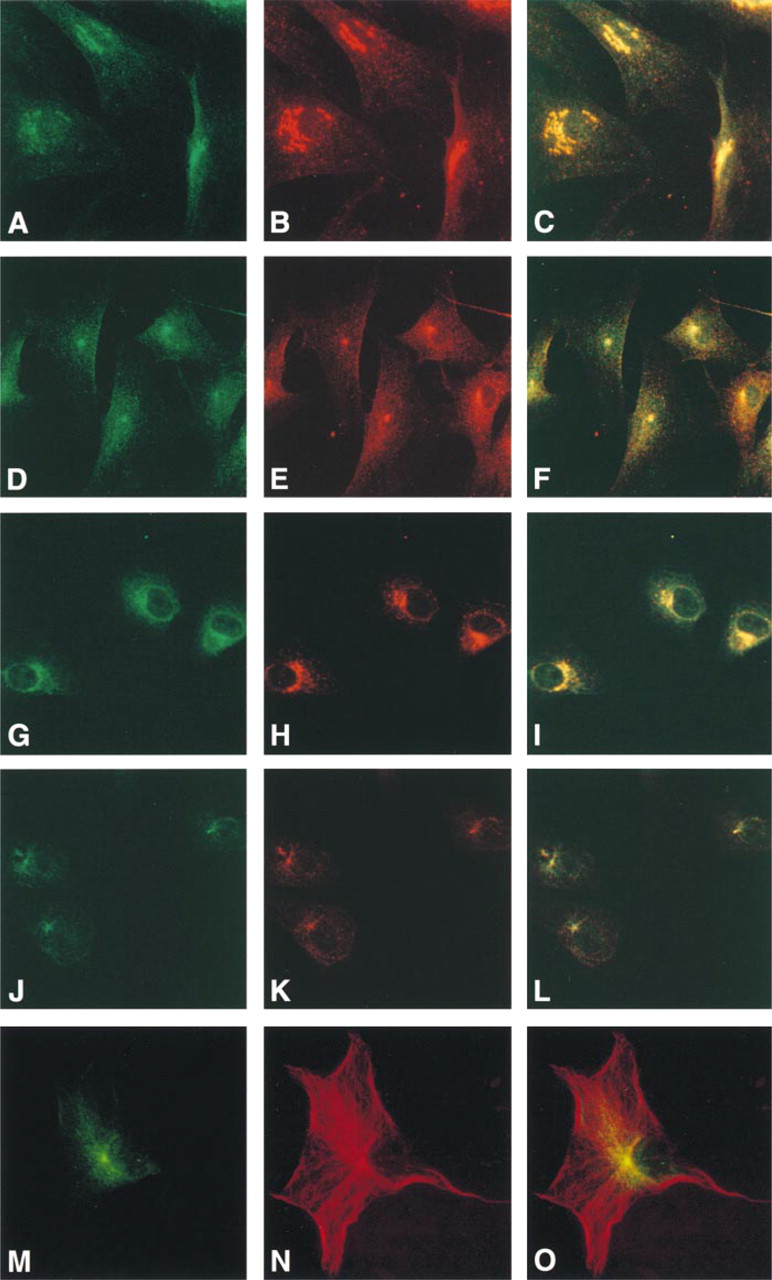
The effect of BFA on the subcellular localization of ocrl1 in fibroblasts (
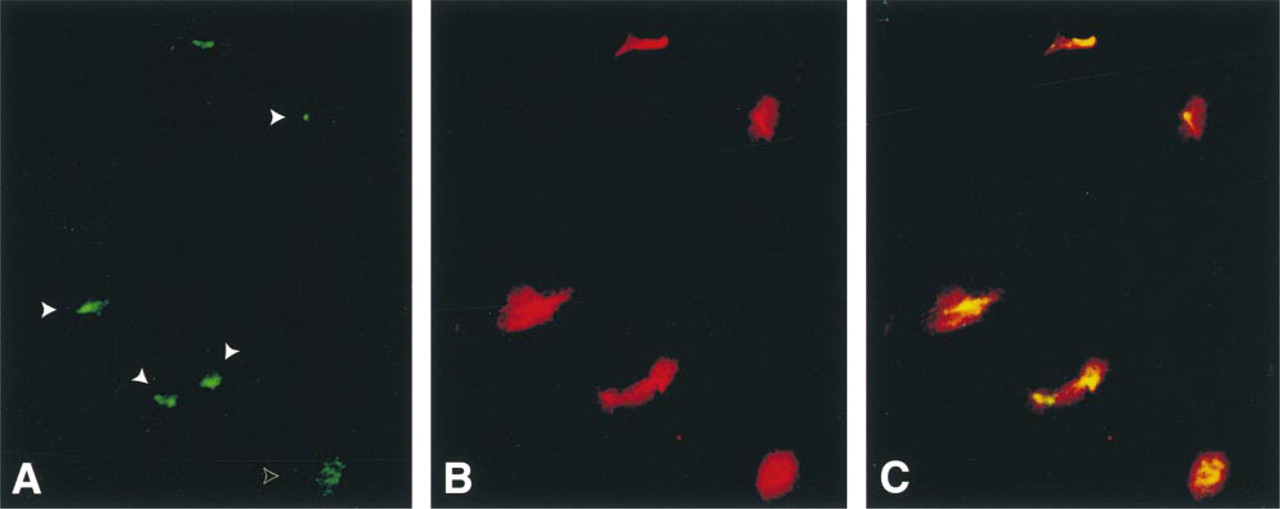
The effect of BFA on the subcellular localization of ocrl1 and lamp1 in NHK cells. After a 60-min incubation with BFA, ocrl1 staining (green) (
We first studied the localization of ocrl1 by immunofluorescence experiments with several markers. In fibroblasts and Vero cells, ocrl1 immunostaining co-localized with that of the TGN protein γ-adaptin. Furthermore, ocrl1 and lysosomal marker localizations were nonoverlapping, making the Golgi localization of ocrl1 clear. In human kidney epithelial cells (NHK), we likewise observed co-localization of ocrl1 and γ-adaptin, which is in contrast to a previous report (Zhang et al. 1998). However, there was also an intersection between ocrl1 and lamp1 staining in the NHK cells. This overlap among γ-adaptin, lamp1, and ocrl1 staining in immunofluorescence experiments led us to the conclusion that this method was inadequate for localization of ocrl1 in NHK cells and that additional methods had to be employed.
To further investigate the localization of ocrl1 in NHK cells, in which the co-localization staining patterns were ambiguous, subcellular fractionation experiments were done. Ocrl1 was concentrated in the Golgi/microsomal fraction as was the TGN protein, γ-adaptin, whereas lamp1 was concentrated in the mitochondrial/lysosomal fraction. The presence of a smaller amount of ocrl1 and γ-adaptin in the mitochondrial/lysosomal fraction may represent limited TGN material in that fraction. The presence of a small percentage of lamp1 in the Golgi/microsomal fraction is expected because electron microscopic studies have found that about 10-30% of lamp1 is present in the TGN (Chen et al. 1985; Geuze et al. 1988; Griffiths et al. 1988, 1990; Honing et al. 1996). The fractionation data clearly demonstrated that ocrl1 and lamp1 are in different subcellular compartments in NHK cells.
To confirm the evidence for a Golgi localization of ocrl1 in NHK cells and to identify the specific Golgi compartment in which ocrl1 was located, we performed perturbation assays using the fungal metabolite BFA. BFA disrupts the Golgi complex and causes transport of Golgi proteins back to the endoplasmic reticulum, resulting in a diffuse cytoplasmic distribution of Golgi proteins. BFA also causes the TGN to tubulate, merge with endosomes, and reversibly condense around the MTOC (Lippincott-Schwartz et al. 1991; Wood et al. 1991; Reaves and Banting 1992). Unlike the effect of BFA on the TGN and endosomes, lysosomes do not condense around the MTOC but instead form tubules that originate from multiple locations throughout the cytoplasm (Lippincott-Schwartz et al. 1991). The different responses of Golgi cisternae, TGN, and lysosomes to BFA provide an established method to distinguish the localization of proteins in these compartments (DeCourcy and Storrie 1991; Ladinsky and Howell 1992). In our experiments, BFA incubation resulted in a redistribution of ocrl1 and γ-adaptin to the MTOC region, whereas no change was detected in lamp1 distribution. On the basis of these dynamic Golgi perturbation experiments, subcellular fractionation studies, and co-localization of ocrl1 with known Golgi proteins, we are able to conclude that ocrl1 is localized to the Golgi in NHK cells, fibroblasts, and Vero cells. The Golgi perturbation experiments more specifically pinpoint the localization of ocrl1 to the TGN.
The overlap between immunofluorescence staining with ocrl1 and lamp1 that we and others (Zhang et al. 1998) observed in NHK cells may be due to several characteristics of these cells. Kidney epithelial cells were observed by us and have been reported previously (Chen et al. 1986) to have a polarized distribution of lysosomes as well as a smaller, more clustered Golgi and a less developed TGN (Sandvig et al. 1991). It has been reported that it is difficult at the light microscopic level to distinguish the specific elements, such as endosomes or lysosomes, in the vicinity of the TGN because of their close proximity (Griffiths et al. 1990). In addition, the more rounded shape of the epithelial cells and the high expression of lamp1 in epithelial cells, particularly kidney, compared with other cell types (Chen et al. 1986) may make it difficult to distinguish TGN and lysosomal staining patterns in these cells. It has also been reported that in rat proximal tubule epithelial cells, there is a prominent M6PR-positive, lysosomal membrane antigen positive late endosomal compartment (Griffiths 1989). If such a compartment were present in human proximal tubule cells, this would confound the distinction between lysosomes and the TGN in these studies.
Ocrl1 is a member of an enzyme family that hydrolyzes the 5-position phosphate from phosphatidylinositol polyphosphate (Suchy et al. 1995; Zhang et al. 1995). Ocrl1 is expressed in almost all tissues examined, with the exception of hematopoietic cells (Olivos-Glander et al. 1995; Jänne et al. 1998), yet a deficiency of this PtdIns(4,5)P2 5-phosphatase, which results in Lowe syndrome (Lowe et al. 1952), affects primarily polarized cells of the lens, kidney, and possibly the brain. The reason why the Lowe syndrome phenotype is restricted to certain epithelial cells is unclear. One possibility is that ocrl1 has a function shared by certain polarized cells. Alternatively, tissues with normal function may have sufficient activity of another PtdIns (4,5)P2 5-phosphatase to compensate for the ocrl1 deficiency. One such enzyme may be inositol polyphosphate 5-phosphatase (inpp5b) (Matzaris et al. 1994). Knockout mice for either ocrl1 or inpp5b are viable and do not have detectable defects in the lens, kidney, or brain. However, ocrl1 and inpp5b double-knockout mice die in the embryonic stage, indicating that these two proteins have some overlapping function (Jänne et al. 1998).
The precise cellular role for ocrl1 is unknown. Furthermore, it is not clear how ocrl1 associates with or is targeted to cell membranes. Sequence analysis of the ocrl1 protein indicates that it has no transmembrane domains nor does it contain a consensus sequence for myristolation or prenylation for membrane attachment. Its association with membranes appears to be weak because ocrl1 is not found with Golgi membranes when fractionation is performed under hypotonic conditions (Suchy and Nussbaum 1998). It is likely to be loosely associated with the TGN or may be a luminal protein. Another member of this family, synaptojanin, appears to be involved in regulating synaptic vesicle budding by hydrolyzing PtdIns(4,5)P2, the required co-factor for PLD (Chung et al. 1997). PLD catalyzes the hydrolysis of phosphatidylcholine to phosphatidic acid, a necessary step for membrane trafficking (Ktistakis et al. 1996). It is possible that ocrl1 plays an analogous role to that of synaptojanin in vesicle budding from the TGN.
The localization of ocrl1 to the TGN is particularly interesting given the special function played by the TGN in epithelial cells. The TGN is the site from which proteins are sorted into vesicles destined for secretory vesicles or lysosomes. In polarized cells, the TGN also serves as the key sorting site for proteins destined for either the apical or basolateral membrane. The localization of ocrl1 to the TGN, its function as a PtdIns(4,5)P2 5-phosphatase, and the restriction of the Lowe syndrome phenotype to certain polarized cells all support the hypothesis that ocrl1 is a second member of the PtdIns 5-phosphatase family that is involved in membrane trafficking, in this case in the TGN. Understanding its role in this specific subcellular compartment may help us to understand the tissue-specific phenotype in Lowe syndrome. It will also prove useful to elucidate more general questions, such as the compensatory capability of other PtdIns (4,5)P2 5-phosphatases and the role of PtdIns (4,5)P2 5-phosphatases in protein sorting in polarized cells.
Footnotes
Acknowledgments
We thank Edwin Arnold, Nancy Theriault, and Darryl Leja for assistance in the preparation of the photomicrographs.
We also thank Drs Deborah Cabin and Christian Lavedan for critical reading of the manuscript.