
Abstract
Objectives:
Genome-wide association studies have identified a significant risk gene, CACNA1C, for schizophrenia. In this study, we comprehensively investigated a large set of CACNA1C single-nucleotide polymorphisms (SNPs) to identify the replicable risk alleles for schizophrenia and explore their biological functions.
Methods:
One Jewish (1044 cases vs 2052 controls), one European (1350 cases vs 1378 controls) and one exploratory African American samples (98 cases vs 20 controls) were analyzed to identify replicable single-nucleotide polymorphism–schizophrenia associations. The regulatory effects of risk alleles on CACNA1C messenger RNA expression were examined. The most robust risk tagSNP (rs1006737) was meta-analyzed on 17 studies (74,122 cases vs 109,062 controls), and associated with the gray matter volumes of seven subcortical structures in 38,258 Europeans, and the surface areas and thickness of 34 cortical regions in 33,992 Europeans and 2944 non-Europeans.
Results:
Forty-seven replicable risk single-nucleotide polymorphisms, including a 20-single-nucleotide polymorphism haplotype block, were identified in our samples (1.8 × 10−4 ⩽ p ⩽ 0.049). This variant block was consistently associated with schizophrenia across four independent Psychiatric Genomics Consortium cohorts (79,645 cases vs 109,590 controls; 2.5 × 10–17 ⩽ p ⩽ 0.017). This block showed significant expression quantitative trait loci in three independent European brain cohorts (5.1 × 10–12 ⩽ p ⩽ 8.3 × 10–3) and could be tagged by the most significant risk single-nucleotide polymorphism rs1006737. The minor allele A of rs1006737 significantly increased risk for schizophrenia across the Jewish and European samples (p = 0.029 and 0.004, respectively), and this association was highly significant in the meta-analysis (p = 1.62 × 10–42). This allele also significantly altered the CACNA1C messenger RNA expression in five brain regions (5.1 × 10–12 ⩽ p ⩽ 0.05), decreased the gray matter volume of thalamus (p = 0.010), the surface area of isthmus cingulate cortex (p = 0.013) and the thickness of transverse temporal and superior temporal sulcus cortexes (0.005 ⩽ p ⩽ 0.043).
Conclusion:
We identified an independent, replicable, functional, and significant risk variant block at CACNA1C for schizophrenia, which could be tagged by the most robust risk marker rs1006737, suggesting an important role of CACNA1C in the pathogenesis of schizophrenia.
Introduction
Schizophrenia is a complex psychiatric disorder characterized by severe emotional, cognitive and social dysfunction. Despite decades of research, the pathogenesis of schizophrenia remains unclear. Many in vivo structural magnetic resonance imaging (MRI) studies have reported reduction in cortical volumes or thickness (TH) in schizophrenia, including the frontal (e.g. pars triangularis (Iwashiro et al., 2012)), temporal (e.g. superior temporal (Ohi et al., 2016)), parietal (precuneus (Forlim et al., 2020)), occipital (e.g. lingual (Yu et al., 2018)) cortices, limbic system (e.g. anterior cingulate (Salgado-Pineda et al., 2014) and thalamus (Andreasen et al., 1994; Byne et al., 2002; Honea et al., 2005; Huang et al., 2017; Konick and Friedman, 2001)) as well as the insula (Shepherd et al., 2012). In contrast, the gray matter volumes (GMVs) of the basal ganglia (e.g. putamen (Hokama et al., 1995) and substantia nigra (Williams et al., 2014)) have been reported to significantly increase in schizophrenia in most published studies.
Substantial evidence has accumulated to suggest strong genetic bases in the pathogenesis of schizophrenia, with the heritability estimated at approximately 83% (Sullivan et al., 2003). Recently, genome-wide association studies (GWASs) have consistently implicated variation within the α-1C subunit of the L-type voltage-gated calcium channel gene, namely Cav1.2 gene (CACNA1C, 12p13.3) as a risk factor of schizophrenia (O’Donovan et al., 2008; Ripke et al., 2013; Stefansson et al., 2009). Located in the membrane of most excitable cells, the alpha-1C subunit of a voltage-dependent calcium channel mediates the influx of calcium ions into the cell upon membrane polarization and regulates intracellular processes such as contraction, secretion, neurotransmission and gene expression. Many variants at CACNA1C have been reported in schizophrenia, some of which are functional and have been significantly and consistently associated with schizophrenia. Among them, rs1006737 is the most significant and best-replicated variant (Ripke et al., 2013; Schizophrenia Psychiatric Genome-Wide Association Study Consortium, 2011; Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). At least six studies with large sample sizes have reported that the minor allele A of rs1006737 significantly increased the risk for schizophrenia in European (Green et al., 2010; Ivorra et al., 2014; Nyegaard et al., 2010) or Asian (Guan et al., 2014; He et al., 2014; Zheng et al., 2014) populations, although some studies of smaller sample sizes did not replicate these results.
In this study, we aimed to comprehensively investigate a large set of CACNA1C single-nucleotide polymorphisms (SNPs) in schizophrenia, in order to identify the replicable associations across independent samples. The most robust risk tagSNP was then meta-analyzed by combining the findings from this and published studies, and its potential biological functions were explored by examining its regulatory effects on CACNA1C messenger RNA (mRNA) expression in the brain, subcortical GMV and cortical surface area (SA) and TH.
Materials and methods
Subjects
Three independent, including one Jewish, one European and one African American, samples were included for gene–disease association analysis. Sample #1 came from the Ashkenazi European data set (dbGaP access number: phs000448.v1.p1), including 1044 Jewish patients with schizophrenia and 2052 healthy Jewish controls. Sample #2 came from the GAIN data set (dbGaP access number: phs000021.v3.p2), including 1350 European patients with schizophrenia and 1378 healthy European controls. Sample #3 came from the non-GAIN data set (dbGaP access number: phs000167.v1.p1), including 98 African American patients with schizophrenia and 20 healthy African American controls.
All subjects were at least 18 years old. Affected subjects met lifetime Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV) criteria for schizophrenia (American Psychiatric Association, 1994). The consensus diagnoses were made by two senior psychiatrists. Patients were excluded if their mental illness was determined to be secondary to a neurological disorder or substance use, or if they had worse than mild mental retardation. Control participants were free from a diagnosis of schizophrenia, schizoaffective disorder, bipolar disorder and major depressive disorder, and from psychotic symptoms, including auditory hallucination and persecutory delusion. All subjects gave written informed consent. Detailed demographic data for these samples have been published in previous studies (Gain Collaborative Research Group et al., 2007; Lencz et al., 2013; Sanders et al., 2008).
Study design
The overall design of this study is illustrated in Figure 1. Briefly, the genotypes were imputed, associated with schizophrenia in the above three samples and referenced to four Psychiatric Genomics Consortium (PGC) samples. The replicable associations across at least two of the above three samples were identified. The regulatory effects of the risk variants on CACNA1C mRNA expression in the brain were examined, to identify potential functional variants. Extracted from these functional risk variants, the most robust risk tagSNP that was most significantly associated with schizophrenia across multiple samples was subject to meta-analysis and evaluated for association with subcortical GMVs and cortical SA and TH.
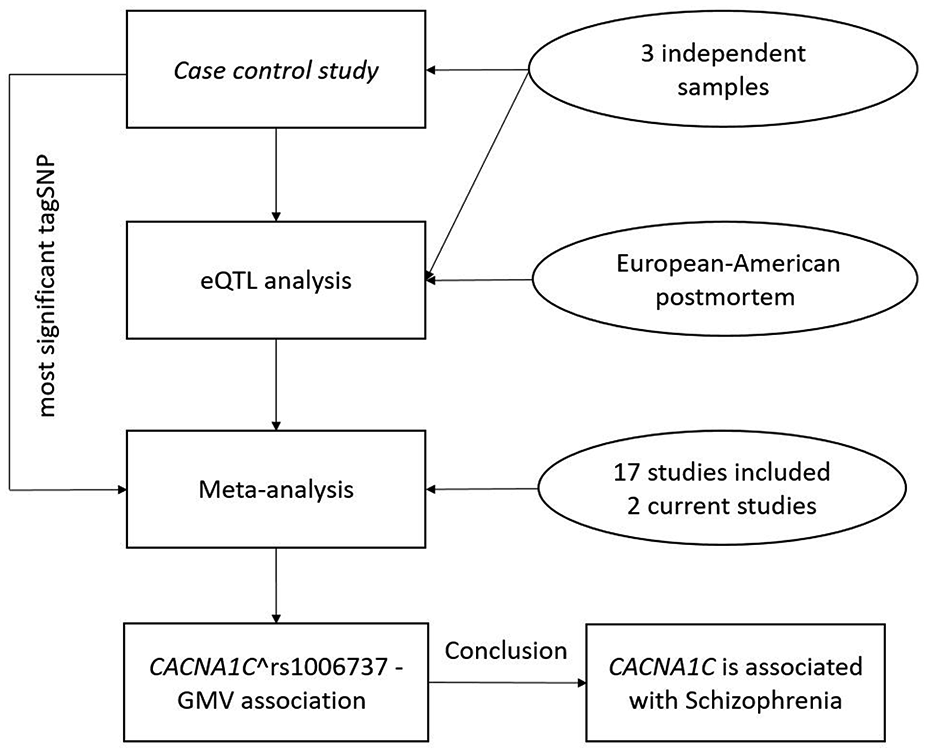
Flowchart of this study. (We did case–control association study between 847 imputed SNPs at CACNA1C and schizophrenia in independent Jewish and European samples [Sample #1: 1044 cases and 2052 controls; Sample #2: 1350 cases and 1378 controls] and one African American sample. A total of 47 SNPs were significantly associated with schizophrenia across at least two samples. To demonstrate the biological function of risk variants, we examined their potential regulatory effects on the CACNA1C mRNA expression in human postmortem brains in a UK European cohort [n = 129] and in a European American cohort [n = 210] using expression quantitative trait locus [eQTL] analysis. We found the most significant tagSNP among these SNPs was rs1006737. We further investigated the result by a meta-analysis combining our results with the findings of previously 15 published studies [total 17 samples: cases = 74,122; controls = 109,062]. The statistical effectiveness of meta-analysis was stronger. Finally, the potential regulatory effects of minor allele A of rs1006737 on the gray matter volumes [GMVs] of seven subcortical structures in a European sample [n = 38,258] and the surface area [SA] and thickness [TH] of 34 cortices in a mixed sample [n = 36,936] were analyzed using multiple linear regression analysis. We identified an independent, replicable, functional, and significant risk variant block at CACNA1C for schizophrenia, which could be tagged by the most robust risk marker rs1006737.)
Genotyping, imputation, data cleaning and single-nucleotide polymorphism–disease association analysis
Sample #1 was genotyped on HumanOmni1-Quad_v1-0_B microarray platform, and Samples #2 and #3 on AFFYMETRIX AFFY_6.0 microarray platform. To make the genetic marker sets consistent across different samples, we imputed the untyped single-nucleotide polymorphisms across the entire CACNA1C region using the same reference panels of 1000 Genome Project and HapMap3 Project data by the program IMPUTE2 (Howie et al., 2009). This entire CACNA1C region starts from Chr12:1948979 (5′-UTR) and ends with Chr12:2677376 (3′-UTR) (Genome Build 36).
Before statistical analysis, we stringently cleaned the phenotype and genotype data, as described in detail previously (Zuo et al., 2012). In brief, subjects with missing diagnosis, missing race or a missing genotype call rate ⩾2% across all SNPs were excluded. Furthermore, we excluded SNPs with an overall missing genotype call rate ⩾2% across all subjects. The SNPs with minor allele frequencies ⩽ 0.01 in either affected or unaffected subjects, or in Hardy–Weinberg disequilibrium (p < 0.001) in unaffected subjects were excluded.
After data cleaning, the allele frequencies of SNPs were compared between individuals with schizophrenia and controls using the Fisher exact test as implemented in the program PLINK. Odds ratios (ORs) were calculated. Hardy–Weinberg equilibrium (HWE) was evaluated using the goodness-of-fit χ2 test. A SNP–disease association with p < 0.05 across at least two samples was taken as a replicable association. Linkage disequilibrium (LD) map of risk variants was analyzed using LDLink to identify risk variant block (r2 > 0.85).
cis-acting expression quantitative trait locus analysis
When a variant is biologically functional, its association with a disease may be true; i.e., the likelihood of false positives of a statistical association is reduced. To demonstrate the biological function of risk variants, we examined their potential regulatory effects on the CACNA1C mRNA expression in human postmortem brains in a UK European cohort (n = 129) (i.e. BRAINEAC data set) (Ramasamy et al., 2014) and in a European American cohort (n = 210) (GTEx Consortium, 2013) using cis-acting expression quantitative trait locus (cis-eQTL) analysis. These subjects were free of neurodegenerative and neuropsychiatric disorders. In the UK European cohort, a total of 10 brain regions were analyzed, including the cerebellar cortex, frontal cortex, hippocampus, medulla, occipital cortex, putamen, substantia nigra, temporal cortex, thalamus and intralobular white matter, and three CACNA1C mRNA isoforms were analyzed, including isoforms #1 (Affymetrix transcript cluster ID [tID]: 3400730; 57 exon probesets; 690,511 bp), #2 (tID: 3401086; 4 exon probesets; 3688 bp) and #3 (tID: 3400695; 1 exon probeset; 138 bp). In the European American cohort, a total of 11 brain regions were analyzed, including the substantia nigra, putamen (basal ganglia), nucleus accumbens (basal ganglia), hippocampus, hypothalamus, frontal Cortex (Brodmann Area or BA9), cerebellum, cerebellar hemisphere, caudate (basal ganglia), anterior cingulate cortex (BA24) and amygdala, and only the longest transcript isoform was analyzed. Normalized mRNA expression levels were compared among different alleles using linear regression analysis.
Meta-analysis
From the risk variants identified from the SNP–disease association analysis, we extracted the most robust risk tagSNP for meta-analysis. This SNP, rs1006737, was biologically functional and most significant in association with schizophrenia across multiple samples, as well as representative of many other SNPs.
Literature search
In EMBASE and PUBMED (as of November 2019) we queried with the searching terms ‘schizophrenia’, ‘CACNA1C’ and ‘rs1006737’ to identify studies appropriate for meta-analysis, without restrictions on language. The retrieved studies were evaluated on the basis of the titles and abstracts to examine the association between CACNA1C gene polymorphism and schizophrenia. If necessary, we also examined the full texts to evaluate whether they were suitable for meta-analysis. Unrelated studies were excluded.
Inclusion and exclusion criteria
Studies met the following criteria to qualify for inclusion in the meta-analysis: (1) evaluation of the relationship between rs1006737 and schizophrenia (2) provision of original data of genotypic and allelic frequencies of cases and controls, (3) inclusion of patients meeting the DSM-IV or the International Classification of Diseases-10 (ICD-10) criteria for schizophrenia and (4) independence from other studies.
Data extraction
For each study included for meta-analysis, two authors extracted the following data: (1) first author and publication year, (2) ethnicity of the subjects, (3) case and control sample sizes, (4) announcement of HWE, (5) OR and 95% confidence interval (CI), (6) p value and (7) genotype information and frequencies of both cases and controls. When these data were not accessible in the published research or supplementary materials, we emailed the correspondence authors and requested for the data.
Analysis of data
In the meta-analysis, the underlying heterogeneity of individual research was tested using the χ2-based Cochran’s Q statistic (Egger et al., 1997), which is a weighted sum of the squares of the deviations of individual OR estimates from the overall estimation. Power analysis was conducted for the entire sample and for different ethnic populations using OR value of 1.20 for the risk allele with a minor allele frequency (MAF) of 0.05, as calculated by Quanto 1.2.4 under the additive model (https://preventivemedicine.usc.edu/download-quanto/). I2 statistic was adopted to estimate the percentage of variation due to heterogeneity other than chance in the study, with extreme, large, moderate and low heterogeneity corresponding to 75–100%, 50–75%, 25–50% and 0–25%, respectively. If the heterogeneity was absent among individual studies, a fixed-effect model was used (Mantel–Haenszel method) to combine each sample and assess the OR value and the corresponding 95% CI. Otherwise, the random-effect model (DerSimonian–Laird method) was used. Subgroup analysis was performed to examine any moderating effect of ancestry (Asian and European). The allele frequency comparison (A and G) was used to calculate the Pooled ORs, and Z score test was used to determine the significance of the pooled ORs. Individual study may have effects on the pooled OR. To investigate such an influence, we performed sensitivity analysis by deleting each study in turn and then recalculating the pooled OR and 95% CI. The primary statistical meta-analysis was conducted with RevMan 5.3 software (www.cochrane.org/revman). The program METAL was employed as an additional tool to produce the exact p values and Z scores. Publication bias was evaluated with Egger’s test (Egger et al., 1997) and shown with Begg’s funnel plots (Begg and Mazumdar, 1994) using Stata 15.1 software (RRID:SCR_007244; Stata Corp). When a two-tailed p value was less than 0.05, we considered it as statistically significant.
Regulatory effects of minor allele A of rs1006737 on the GMVs of subcortical structures
The GMVs of basal ganglia (caudate, putamen, pallidum and accumbens) and limbic system (amygdala, hippocampus and thalamus) in 38,258 European subjects (ENIGMA2 data set) (Hibar et al., 2015; Satizabal et al., 2019) were measured by structural MRI, following a standardized protocol. The GMVs were calculated using the brain segmentation software packages: FIRST or FreeSurfer. All subjects were genotyped using microarray and imputed to the 1000 Genome Project genotype panels. The genetic homogeneity was assessed in each subject using multi-dimensional scaling (MDS).
The potential regulatory effect of minor allele A of rs1006737 on the GMVs was analyzed using multiple linear regression analysis, controlling for age, age2, sex, four MDS components, total intracranial volume and diagnosis (where applicable; most subjects were free of neurodegenerative and neuropsychiatric disorders).
Regulatory effects of minor allele A of rs1006737 on the SA and average TH of cortexes
A total of 36,936 subjects, including 33,992 Europeans (23,909 from 49 ENIGMA cohorts and 10,083 from the UK Biobank) and 2944 non-European participants (eight cohorts) (Grasby et al., 2020), were included in this analysis. Measures of cortical SA and TH of these subjects were derived from in vivo whole brain T1-weighted MRI scans using FreeSurfer. SA and TH were quantified for each subject across the whole cortex and within 34 distinct gyral-defined regions in each brain hemisphere according to the Desikan–Killiany atlas. SA was measured at the gray-white matter boundary, and TH was measured as the average distance between the white matter and pial surfaces. The SA and TH of each cortical region were analyzed separately based on the radial unit hypothesis that different developmental mechanisms promoted SA expansion and TH increases. The total SA and average TH were also computed.
The potential regulatory effects of minor allele A of rs1006737 on a total of 70 traits (total SA, average TH and the SA and TH of 34 cortical regions averaged across right and left hemispheres) were analyzed by exploring the associations between this allele and the traits. These associations were analyzed by multiple linear regression analyses, adjusting for the effects of sex, linear and nonlinear age effects, interactions between age and sex, ancestry (the first four MDS components), diagnostic status (when the cohort followed a case–control design), MRI acquisition orientation, dummy variables for scanner (when multiple scanners were used at the same site) and the global measure (total SA or average TH).
Results
An independent variant block was significantly associated with schizophrenia across multiple samples/cohorts
A total of 847 imputed SNPs were analyzed, of which 47 SNPs were significantly associated with schizophrenia across at least two independent samples (1.8 × 10–4 ⩽ p ⩽ 0.049; Figure 2). LD map showed that most of these risk SNPs were clustered in two independent (r2 > 0.85) haplotype blocks (Figure 2). The 20 SNPs within the first haplotype block (from rs10848627 to rs1006737) were highly significantly associated with schizophrenia across at least three of the four PGC cohorts (2.5 × 10–17 ⩽ p ⩽ 0.017; Table 1); however, the 20 SNPs within the second haplotype block (from rs4765966 to rs2302729) were not (all p > 0.05; data not shown). cis-eQTL analysis showed that the SNPs within the first block had significant eQTL signals across three independent European cohorts (5.1 × 10–12 ⩽ p ⩽ 8.3 × 10–3; Table 1), but those in the second block did not (all p > 0.05; data not shown).
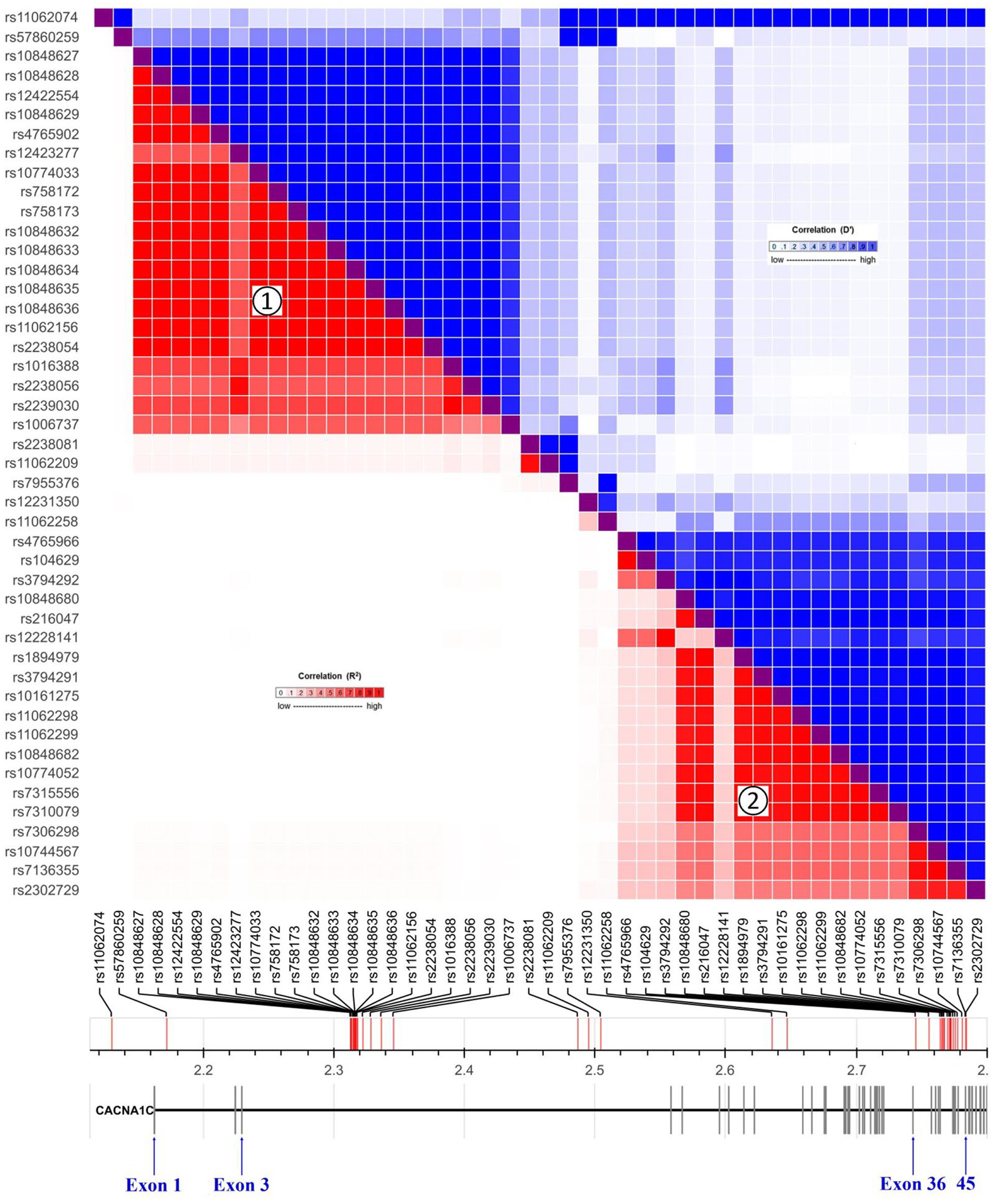
LD map of risk variants for schizophrenia. A total of 47 replicable risk SNPs for schizophrenia were identified across the CACNA1C, including 20 SNPs within the first haplotype block ①, 20 SNPs within the second haplotype block ②, and other seven independent SNPs. Red squares indicated r2 values, and blue squares indicated D′ values. This LD map was generated by LDLink.
p values for SNP–schizophrenia and mRNA associations in the first haplotype block.
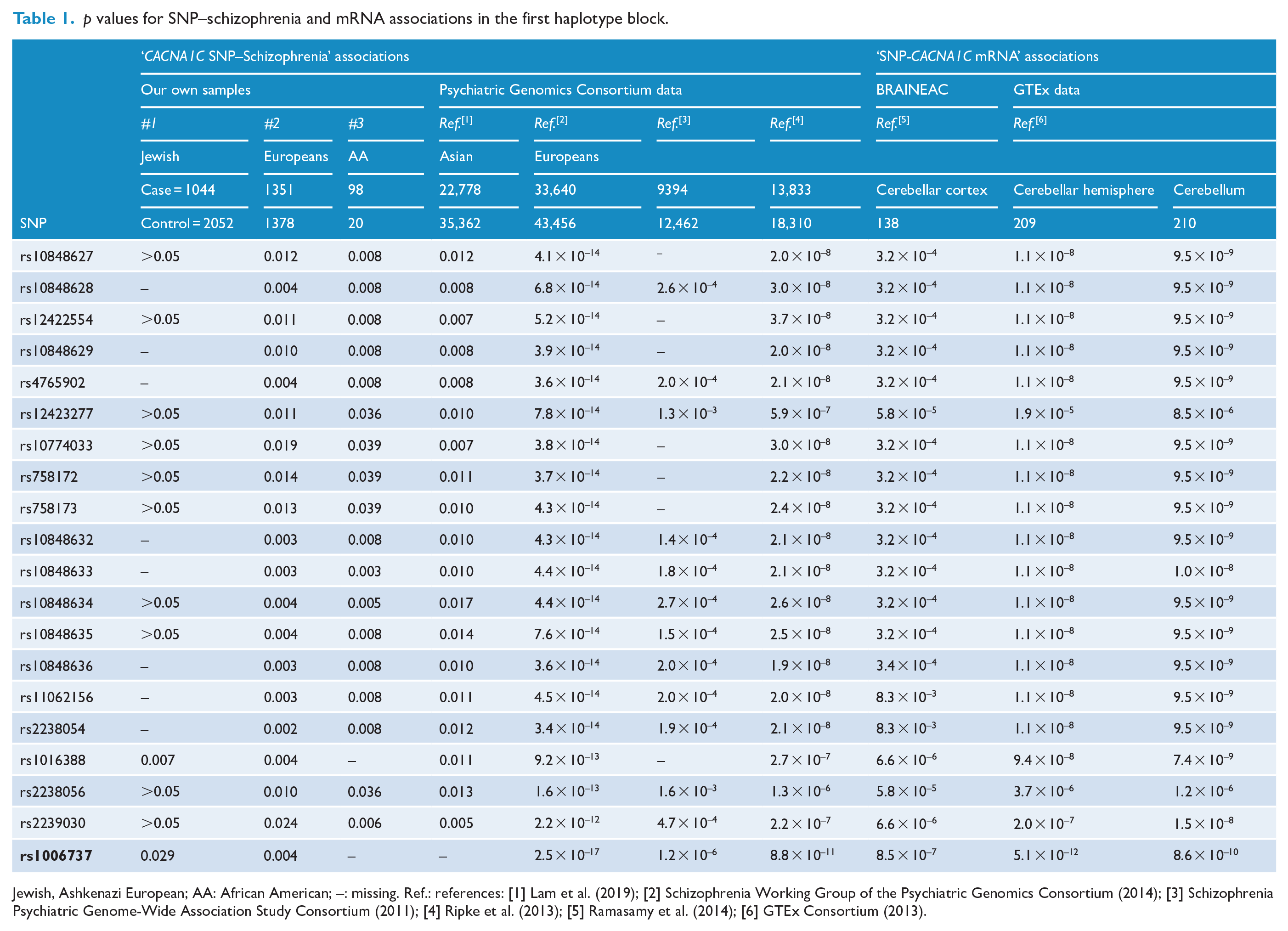
Jewish, Ashkenazi European; AA: African American; –: missing. Ref.: references: [1] Lam et al. (2019); [2] Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014); [3] Schizophrenia Psychiatric Genome-Wide Association Study Consortium (2011); [4] Ripke et al. (2013); [5] Ramasamy et al. (2014); [6] GTEx Consortium (2013).
The most significant tagSNP among the first block was rs1006737. Its genotype frequency distributions both in cases and controls were in accordance with the HWE (p > 0.05). It was significantly associated with schizophrenia in Samples #1 and #2 (p = 0.029 and 0.004, respectively; Tables 1 and 2), and highly significantly associated with schizophrenia across the three European PGC cohorts (2.5 × 10–17 ⩽ p ⩽ 1.2 × 10–6; Tables 1 and 2). The minor allele A increased risk for schizophrenia in all of these five samples/cohorts (OR > 1; Table 2 and Figure 3).
Characteristics of included studies for meta-analysis of minor allele A of rs1006737.
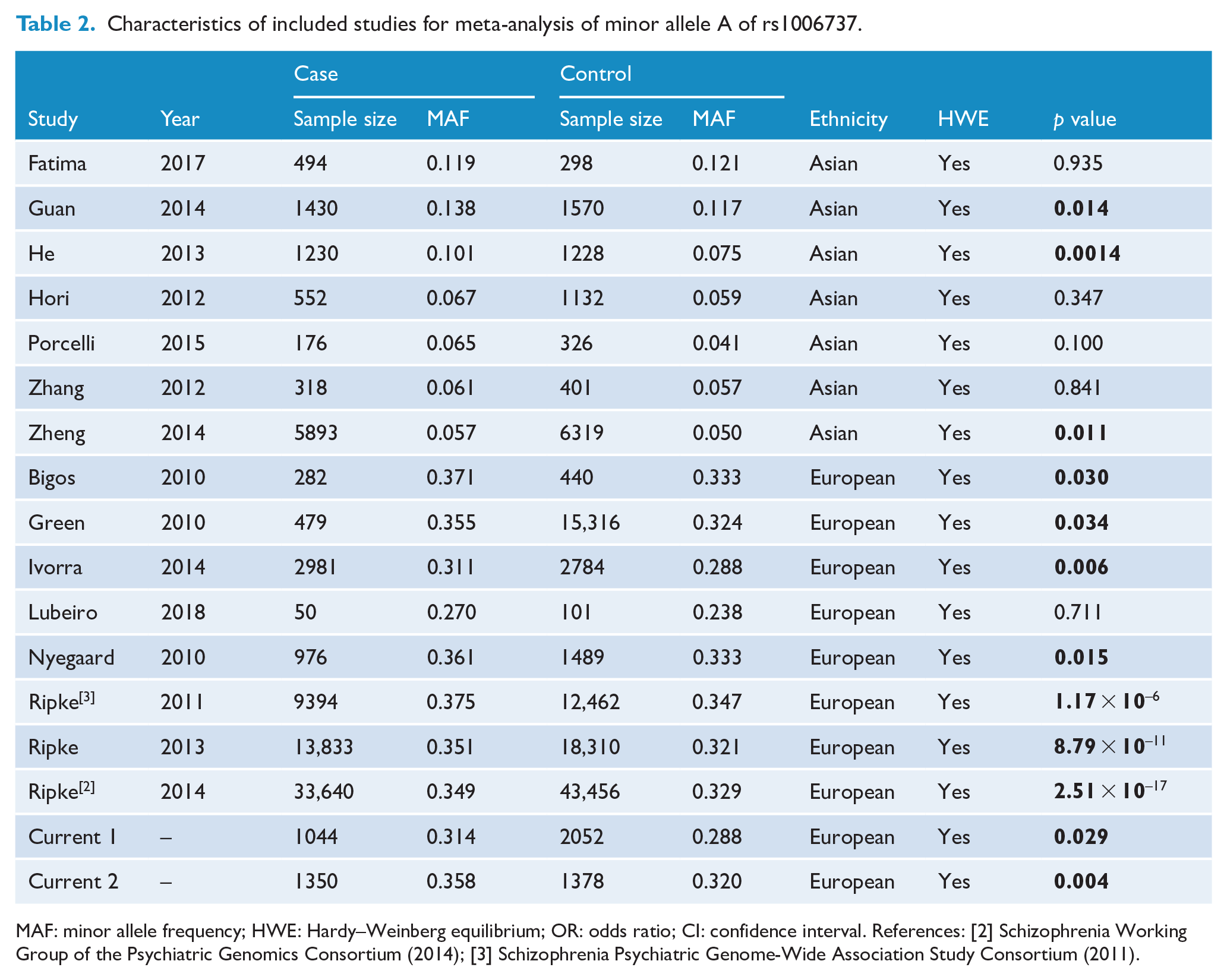
MAF: minor allele frequency; HWE: Hardy–Weinberg equilibrium; OR: odds ratio; CI: confidence interval. References: [2] Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014); [3] Schizophrenia Psychiatric Genome-Wide Association Study Consortium (2011).
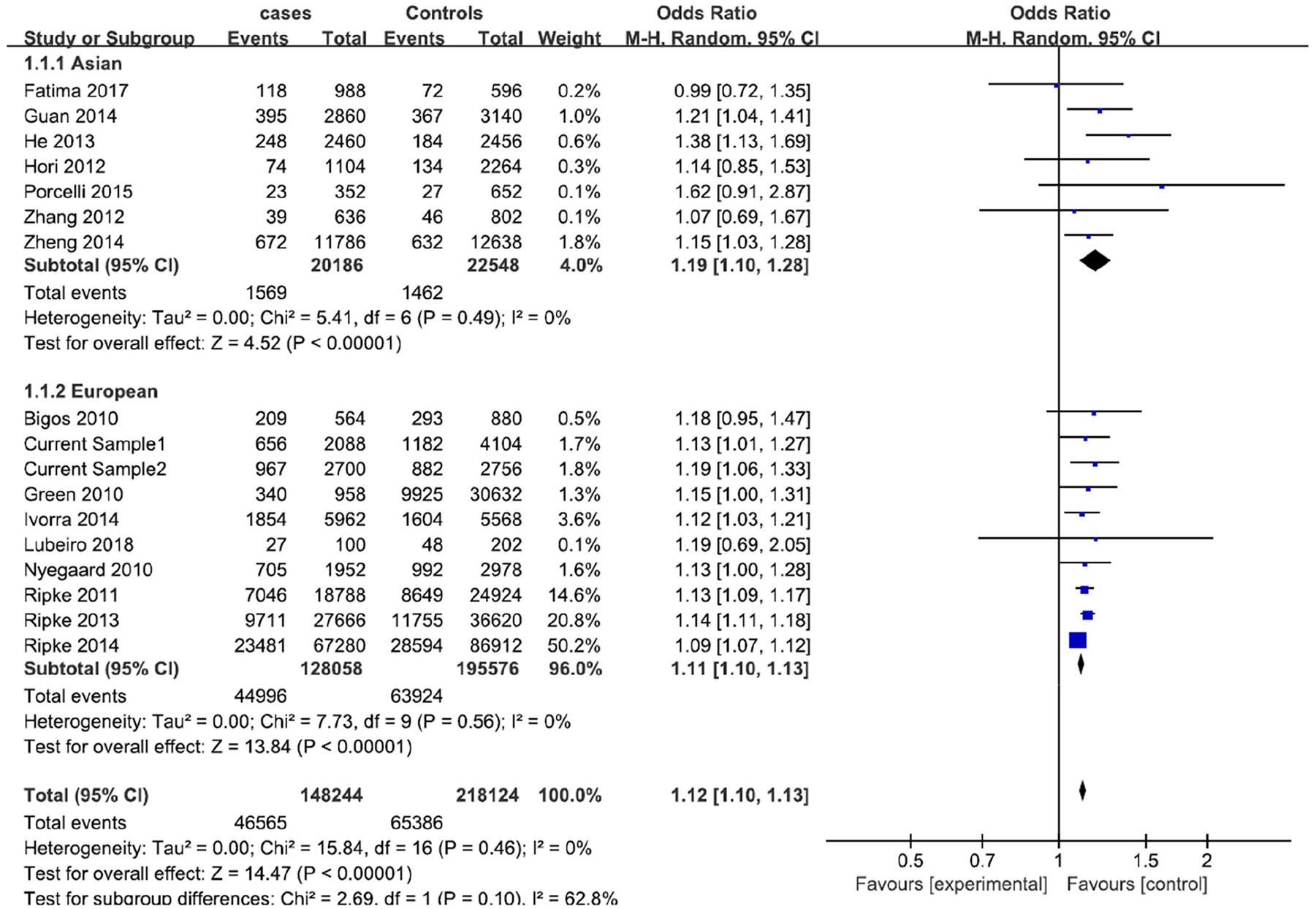
Meta-analysis for the association of allele A of rs1006737 with schizophrenia in the Asian, European and combined populations.
cis-eQTL analysis of rs1006737
The minor allele A ( f < 0.5) of rs1006737 significantly increased the CACNA1C mRNA expression in the cerebellar hemisphere (normalized effect size [NES] = 0.63; p = 5.1 × 10–12), cerebellum (NES = 0.54; p = 8.6 × 10–10), putamen (NES = 0.06; p = 0.05) and substantia nigra (NES = 0.21; p = 0.02) (GTEx data; Table 3), but decreased the expression in the cerebellar cortex (BRAINEAC data; t < 0; p = 8.5 × 10–7 for isoform #1 and p = 9.9 × 10–3 for isoform #2; Table 3).
Associations between minor allele A of rs1006737 and mRNA expression or GMV.

Only the longest isoform of CACNA1C transcripts is listed. GMV: gray matter volume of brain region; n: sample size; NES: normalized effect size. Ref.: references: [5] Ramasamy et al. (2014); [6] GTEx Consortium (2013); [7] Satizabal et al. (2019).
Meta-analysis of associations between rs1006737 and schizophrenia
Power analysis and HWE assessment
We performed a power analysis to detect significant allelic association. The entire sample (including the current independent Jewish and European samples) with 74,122 cases and 109,062 controls was sufficient to detect a statistical difference with a power of near 100%. The power for the Asian (case = 10,093; control = 11,274) and European (case = 64,029; control = 97,788) sample alone was near 99% and 100%, respectively. The genotype frequency distribution of each group was consistent with HWE.
Meta-analysis of studies of rs1006737
Seventeen (including the current two independent samples) studies meeting the inclusion and exclusion criteria were included in the meta-analysis. Figure S1 shows a flowchart of the literature search and study selection process. A total of 74,122 schizophrenia cases and 109,062 healthy controls were included in the meta-analysis. These studies are described in Table 2.
Meta-analysis results
When all 17 samples were included (case = 74,122; control = 109,062), the association between schizophrenia and CACNA1C^rs1006737 allele frequency was highly significant (METAL: Z = 13.67, p = 1.62 × 10–42; RevMan: Z = 14.47, p < 10–5, OR = 1.12, 95% CI: 1.10–1.13) under the fixed effects model. There was no heterogeneity in the included studies (p = 0.46, I2 = 0.0%). The meta-analysis forest plot is illustrated in Figure 3.
We further analyzed the studies separately for Asian and European populations. Because no heterogeneity was observed in the Asian (p = 0.49, I 2 = 0.0%) or European (p = 0.56, I2 = 0.0%) studies, we used the fixed effects model in the meta-analysis. The meta-analysis of the seven Asian samples including 10,093 patients and 11,274 healthy controls showed a significant association between CACNA1C^rs1006737 and schizophrenia (METAL: Z = 4.44, p = 9.09 × 10–6; RevMan: Z = 4.52, p < 10–5, OR = 1.19, 95% CI: 1.10–1.28). The meta-analysis of the 10 (including the current 2) European samples, including 64,029 cases and 97,788 controls, also showed a highly significant association between CACNA1C^rs1006737 and schizophrenia (METAL: Z = 12.94, p = 2.61 × 10–38, RevMan: Z = 13.84, p < 10–5, OR = 1.11, 95% CI: 1.10–1.13).
Sensitivity analysis
We conducted ‘leave-one-out’ sensitivity analysis through omitting a single research at a time to assess the impact of individual studies on the meta-analysis. The results showed that the meta-analysis using the remaining samples still yielded strong associations and were robust. The details of sensitivity analysis are shown in Table S1.
Publication bias
Neither Egger’s test nor Begg’s test indicated any evidence of publication bias in the total samples (t = −0.23, p = 0.820). Begg’s funnel plot is shown in Figure S2.
The minor allele A of rs1006737 decreased the thalamus GMV
The allele A was significantly associated with lower GMV of the thalamus in Europeans (Z = −2.58; p = 0.010; Table 3). No regulatory effects on other subcortical structures were found (p > 0.05).
The minor allele A of rs1006737 significantly regulated the SA and TH of cortexes
The minor allele A of rs1006737 increased (β > 0) the SA of paracentral, pars triangularis and parahippocampal cortices (0.008 ⩽ p ⩽ 0.033), but decreased (β < 0) the SA of isthmus cingulate cortex (p = 0.013) (Table 4). In addition, this allele increased (β > 0) the TH of precuneus (p = 0.035), but decreased (β < 0) the TH of transverse temporal cortex and superior temporal sulcus (0.005 ⩽ p ⩽ 0.043) (Table 4). No regulatory effects on other regions were found (p > 0.05).
Associations between minor allele A of rs1006737 and cortical SA and TH.

SA: surface area; TH: thickness; Eur: Europeans; UKBB: Europeans from UK Biobank.
Discussion
We tested a large set of CACNA1C SNPs and identified 47 replicable risk SNPs for schizophrenia including two independent haplotype blocks. The first 20-SNP haplotype block showed significant eQTL signals and could be tagged by the most significant risk SNP, rs1006737. The minor allele A of rs1006737 significantly increased risk for schizophrenia across at least five independent European samples, in accordance with previous reports (Bigos et al., 2010; Green et al., 2010; Jiang et al., 2015; Nyegaard et al., 2010; Ripke et al., 2013; Zheng et al., 2014), but not significantly in the African sample, which was probably due to the small sample size.
To further validate the association between the minor allele A of rs1006737 and schizophrenia, we conducted a largest-scale meta-analysis by combining our results with those published previously (74,122 cases vs 109,062 controls). The results demonstrated a highly significant association with a p = 1.62 × 10–42. With the samples stratified by race (i.e. Asian and European), the associations remained significant. These findings thus confirmed previous reports with relatively large sample size in Asian and European populations (Bigos et al., 2010; Guan et al., 2014; He et al., 2014; Ivorra et al., 2014; Nyegaard et al., 2010; Ripke et al., 2013; Zheng et al., 2014).
Likely due to ethnic genetic heterogeneity, the findings associating genetic polymorphisms with the susceptibility of complex human diseases have not always been replicated. Since rs1006737’s MAFs varied much across populations, ranging from 0.288 (European populations) to 0.050 (Asian populations), we conducted heterogeneity analysis. Consistent with previous studies (Jiang et al., 2015; Zheng et al., 2014; Zhu et al., 2019), our meta-analysis indicated no significant heterogeneity between Asian and European populations. We also tested the heterogeneity of European and Asian populations separately to examine potential heterogeneity among individual populations with similar ethnicity. Likewise, we did not find heterogeneity, even if the MAFs were different among populations. To a certain extent, the results of these meta-analyses may explain the inconsistent genome-wide association findings published so far and suggest a role of risk allele of rs1006737 in the pathophysiology of schizophrenic across different ethnic populations.
Publication bias is well known in clinical literature (Begg and Mazumdar, 1994; Egger et al., 1997). Positive results are published more frequently and faster, and are cited more widely. It should be noted that we have included the studies of negative results in the current meta-analysis (Porcelli et al., 2015). On the other hand, findings of negative results may be more common and not restricted to those that have been published. Therefore, we investigated this issue and the results confirmed no significant publication bias in the meta-analysis.
Furthermore, using eQTL analysis, we found that the minor allele A of rs1006737 highly significantly increased the CACNA1C mRNA expression in the cerebellum, and modestly in the putamen and substantia nigra (GTEx data), but decreased the expression in cerebellar cortex (BRAINEAC data). This was consistent with previous findings that the cerebellum and striatum, respectively, exhibited the highest and second highest levels of notable CACNA1C mRNA splicing transcript diversity in terms of abundance and identity (‘isoform’) (Clark et al., 2020). rs1006737 had more significant regulatory effects on the longer transcript isoforms than the shorter ones. The positive regulatory effect on the CACNA1C mRNA expression reduced the likelihood of false positives of SNP–schizophrenia associations, because biologically functional variants are more likely to be true in its association with a disease (Nicolae et al., 2010).
We also found that the allele A of rs1006737 significantly decreased the GMV of thalamus, the SA of isthmus cingulate cortex and the TH of transverse temporal cortex and superior temporal sulcus, but increased the SA of paracentral, pars triangularis and parahippocampal gyri and the TH of precuneus. The thalamus is a critical component of the ‘cortico-basal ganglia-thalamo-cortical’ loop, which supports a wide array of cognitive and affective functions. Studies have associated reduction of thalamus volume with the impairment in social functioning (Koshiyama and Kasai, 2019) and delusions (Huang et al., 2017) in patients with schizophrenia. The transverse temporal gyrus is the first cortical structure to process incoming auditory information and its volume reduction may contribute to auditory hallucination in patients with schizophrenia (Mondino et al., 2020). The isthmus cingulate cortex and superior temporal sulcus are assumed to be implicated in visuospatial orientation and social perception, respectively. Both transverse temporal gyrus and superior temporal sulcus are lateral to the superior temporal gyrus that is implicated in auditory processing, language and social cognition too (Bigler et al., 2007). Our findings that the allele A of rs1006737 significantly decreased the TH of both superior temporal sulcus and transverse temporal gyrus were consistent with the previous report that this allele significantly decreased CACNA1C mRNA expression in the superior temporal gyrus (Eckart et al., 2016). Thus, these downregulatory effects of the allele A of rs1006737 on these cortical and subcortical structures might be involved in the pathogenesis of schizophrenia. In contrast, the implications of increased SA of paracentral cortex, pars triangularis and parahippocampal gyrus, as well as TH of the precuneus remained to be clarified.
A number of additional issues need to be considered for the study. First, we did not examine the rs1006737–schizophrenia association in Asians in our own samples. To address this issue, we performed meta-analysis to combine all of the previously published association studies in Asians, and found that the association was also significant in Asians. Second, although the largest, the sample used in the meta-analysis was susceptible to population stratification. The subjects in some of the studies (e.g. Zheng et al., 2014) had been shown to form a homogeneous cluster of unobservable substructures because of recruiting from the same geographic area. For this reason, we performed sensitivity analysis. After excluding these studies, the results remained significant.
CACNA1C has been identified as a cross-disorder risk gene for schizophrenia, bipolar disorder, major depressive disorder and autism (Dedic et al., 2018). If this set of risk variants identified in this study was shared by the susceptibility to these major psychiatric disorders, it may support the common genetic factor underling the overlapping symptomatology of these disorders. Otherwise, if different sets of variants confer to the risk for different disorders, it may support the heterogeneous roles of CACNA1C variants in different phenotypes. This hypothesis warrants further examination.
As another further research direction, functional studies using animal models could follow up in the future. For example, rs1006737 could be precisely knocked out in the mice using CRISPR/Cas9 genome editing technology, to explore the functional effects of this variant on mRNA and protein expression in the brain, regional volumetric alteration and schizophrenia-like behaviors.
In summary, we identified an independent, replicable, functional and significant risk variant block at CACNA1C for schizophrenia in Europeans and Asians, which could be tagged by the most robust risk marker rs1006737, suggesting an important role of CACNA1C in the pathogenesis of schizophrenia.
Supplemental Material
sj-docx-1-anp-10.1177_00048674211009595 – Supplemental material for An independent, replicable, functional and significant risk variant block at intron 3 of CACNA1C for schizophrenia
Supplemental material, sj-docx-1-anp-10.1177_00048674211009595 for An independent, replicable, functional and significant risk variant block at intron 3 of CACNA1C for schizophrenia by Zuxing Wang, Wenzhong Chen, Yuping Cao, Yikai Dou, Yingmei Fu, Yong Zhang, Xingqun Luo, Longli Kang, Na Liu, Yun Stone Shi, Chiang-shan R Li, Yifeng Xu, Xiaoyun Guo and Xingguang Luo in Australian & New Zealand Journal of Psychiatry
Footnotes
Acknowledgements
The authors thank NIH GWAS Data Repository, the Contributing Investigator(s) who contributed the phenotype and genotype data from his or her original study (e.g. Drs. Lencz, Darvasi, Gejman, Levinson, Goldstein, Stefansson, Collier, Sklar, Collins, Cloninger) and the primary funding organization that supported the contributing study. The data sets used for the analyses described in this manuscript were obtained from dbGaP at ![]() . The dbGaP accession numbers include phs000021.v3.p2, phs000448.v1.p1 and phs000167.v1.p1.
. The dbGaP accession numbers include phs000021.v3.p2, phs000448.v1.p1 and phs000167.v1.p1.
Author Contributions
Z.W., X.G. and X.L. wrote this article; Y.F., W.C., Y.C., Y.X., Y.D., Y.Z., N.L., Y.S.S. did the statistical analysis; X.G., C-s.L., X.L., N.L. and Y.S. edited this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This study is also funded by the National Natural Science Foundation of China (81201057), Shanghai Municipal Health Bureau Project (20124109), Chinese Medical Association, Psychiatry—Servier Youth Research Fund, Shanghai Mental Health Center international cooperation project (2013-) and Shanghai Municipal Center for Mental Health Clinical Research Program. Funding and other supports for phenotype and genotype data were provided through the National Institutes of Health (NIH) (RC2 MH089964, R01 MH084098, R01 MH67257, R01 MH59588, R01 MH59571, R01 MH59565, R01 MH59587, R01 MH60870, R01 MH59566, R01 MH59586, R01 MH61675, R01 MH60879, R01 MH81800, U01 MH46276, U01 MH46289 U01 MH46318, U01 MH79469 and U01 MH79470), the North Shore—LIJ Health System Foundation and the Hebrew University Genetic Resource.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.