
Abstract
Background:
With an estimated 80% heritability, molecular genetic research into schizophrenia has remained inconclusive. Recent large-scale, genome-wide association studies only identified a small number of susceptibility genes with individually very small effect sizes. However, the variable expression of the phenotype is not well captured in diagnosis-based research as well as when assuming a ‘heterogenic risk model’ (as apposed to a monogenic or polygenic model). Hence, the expression of susceptibility genes in response to environmental factors in concert with other disease-promoting or protecting genes has increasingly attracted attention.
Method:
The current review summarises findings of microarray gene expression research with relevance to schizophrenia as they emerged over the past decade.
Results:
Most findings from post mortem, peripheral tissues and animal models to date have linked altered gene expression in schizophrenia to presynaptic function, signalling, myelination, neural migration, cellular immune mechanisms, and response to oxidative stress consistent with multiple small effects of many individual genes. However, the majority of results are difficult to interpret due to small sample sizes (i.e. potential type-2 errors), confounding factors (i.e. medication effects) or lack of plausible neurobiological theory.
Conclusion:
Nevertheless, microarray gene expression research is likely to play an important role in the future when investigating gene/gene and gene/environment interactions by adopting a neurobiologically sound theoretical framework.
Keywords
Introduction
Schizophrenia is a low prevalence disorder affecting 0.7% to 1.4% of the population in a wide variety of geographic regions across the world (Jablensky et al., 1992). The heritability is high and estimated to contribute up to 83% (Canon et al., 1998; Lewis and Lieberman, 2000). However, the molecular genetics have not been identified despite recent large-scale genome-wide association studies involving up to 27,000 individuals (International Schizophrenia Consortium, 2009; Shi et al., 2009; Stefansson et al., 2009). The largest genome-wide association study into schizophrenia to date confirmed two (6p21.32-p22.1 and 18q21.2) and identified five novel loci (1p21.3, 2q32.3, 8p23.2, 8q21.3 and 10q24.32-q24.33) when investigating 51,695 individuals (Ripke et al., 2011). A main obstacle is the clinical phenomenology of schizophrenia which overlaps with other psychotic, affective and personality disorders. Hence, a ‘heterogenic risk model’ (as apposed to a monogenic or polygenic model) probably best accounts for the complex clinical syndromes and diagnostic subtypes of schizophrenia as well as the variable expression of symptoms during the course of illness (Galter et al., 2007; Jablensky, 1995).
Biomarkers or ‘endophenotypes’ of the disorder have been proposed to facilitate more targeted neurogenetic research (McGuffin et al., 1987). Promising findings are emerging from neurocognitive, neurophysiological and brain imaging research revealing a pattern of regional grey and white matter pathology that is also associated with the defining clinical, neurocognitive and pathophysiological features of the disorder (Andreasen et al., 2007; Tsuang et al., 2005).
Within a neurodevelopmental framework, the evolving phenotype is dependent on environmental factors which are impacting on the normal trajectory of brain development. This ‘gene by environment interaction’ is captured by gene expression; that is, the transcription of a gene’s DNA information into a mRNA copy to direct the synthesis of proteins via the genetic code. The transcription process itself reflects a major cellular mechanism of adaptation of cell function in its immediate environment at any given point in time. This also implies that gene expression data can capture mal-adaptation by identifying a signature of over and/or under-expressed quantities of RNA that is associated with a disease. Hence, studies into gene expression have gained substantial interests in schizophrenia research since it holds the promise of identifying a signature of a pathological mechanism at the molecular level.
The current review summarises some of the findings of microarray gene expression research with relevance to schizophrenia as they emerged over the past decade. Given the nature of this new technology – which is often applied without strong theoretical considerations – the studies will be reviewed by the sources of tissue (i.e. port mortem, peripheral tissues and animal models) rather than a theoretical framework. Nevertheless, the authors will attempt to discuss the findings in the context of patho-aetiological models as well as study limitations.
Microarray gene chip methodology
With recent technological advances it is now possible to analyse the expression of the entire genome (or ‘transcriptome’) in a single experiment. Historically, research has been limited to small numbers of genes when using, for instance, northern blot, in situ hybridisation, or polymerase chain reaction (PCR) methods (Deb et al., 1999; Yurov et al., 2001). On the other hand, ‘microarrays’ provide a platform for the simultaneous screening of a large number of genes from a variety of in vivo and post mortem tissues, including the examination of gene expression in single cells.
Microarrays were originally developed from ‘DNA microarrays’ containing complementary DNAs (cDNAs) for particular genes spotted onto nylon membranes that were then used in combination with radioactive probes to determine the levels of mRNA for those genes. In the mid-1990s, cDNAs were spotted onto glass microscope slides and this, coupled with fluorescent probes and improvements in scanning technologies, saw the development of DNA microarrays that significantly increased the number of genes that could be analysed in one single experiment. Over the past decade these technologies rapidly developed and expanded to allow the detection and characterisation of many genetic features, including single nucleotide polymorphisms (SNPs), methylation status, alternative splicing of genes, the actual gene sequence, the actual number of gene copies referred to as copy number variations (CNVs; inherited, somatic, or de novo), as well as, for instance, loss of heterozygosity in cancer cells.
Most of the microarrays are produced by photolithographic in situ oligonucleotide synthesis on a solid surface (such as a glass microscope slide), microscopic beads and plastic- or silicon-based chips, and all are sometimes referred to as ‘gene chips’. Today’s microarrays can feature a density > 750,000 probes for genes or features per array. While this technology now provides an efficient way of screening vast numbers of genes, it also poses the risk of false positive findings due to the high number of tests (i.e. each individual gene comparison constitutes a single statistical test), which are commonly performed without a priori hypothesis. However, this method certainly has its place when aiming for discovery as long as the results are independently reproducible and the findings are critically evaluated with reference to other data (often post hoc) and in the context of existing theoretical models.
One of the early analytic approaches for DNA microarrays was based on computing dendrograms of pairwise comparisons of gene expression (e.g. an individual index patient versus a matched healthy control subject) for a number of such paired comparisons within a single experiment. The resulting dendrogram represents the pattern of gene expression differences between patient and control data. Importantly, the measure taken here is the relative difference of gene expression in each individual pair as measured by a dye marker (e.g. patient sample red and control sample green) which indicates the relative expression difference within the red-green spectrum; that is, a higher reading in the red spectrum indicates a higher expression in the patient sample relative to the matched control sample by x-fold and vice versa. Pooled samples (i.e. establishing a relative measure for the average of a sample) have also been used; however, this lacks some control over potentially confounding variables, such as clinical features, duration of illness, age, sex, medication status, post mortem interval, for example, which can be used as an independent variable (e.g. old versus young, male versus female, etc.) when computing dendrograms.
Earlier methods include Serial Analysis of Gene Expression or SAGE, introduced by Velculescu et al. (1995) to describe the transcriptome in diseases like cancer in a wide range of organisms. More recent technological advances also allow for absolute expression measures which, in turn, can be used to calculate correlations. For instance, when comparing U133Plus2.0 Affymetrix microarray gene expression data of renal allograft biopsies with reverse transcriptase PCR (RT-PCR) data, Allanach et al. (2008) reported significant correlations of most RT-PCR probes with the corresponding microarray data. Nevertheless, microarray data should always be confirmed by another method, such as real-time RT-PCR, which offers a well-accepted quantitative measure of gene expression.
With the availability of different suppliers of microarray chips (e.g. Affymetrix, Illumina, Agilent, etc.), inter-laboratory and cross-platform reliability testing has become possible. Such testing suggests excellent reliability between laboratories and chips (Mao et al., 2007). However, when comparing the two high-resolution microarray platforms using DNA copy number variance as an indicator on a collection of established human melanoma cell lines, Greshock et al. (2007) reported that Affymetrix’s single nucleotide polymorphism microarrays offered better detection of dose-dependant changes in gene expression (also replicated by Mao et al., 2007) while Agilent’s 60-mer oligonucleotide microarray with probe design optimised for genomic hybridization seems to offer higher sensitivity and specificity (Greshock et al., 2007; Agilent Technologies Inc, 2001).
As such, microarray gene chip technology has matured and developed into an important tool of molecular genetic research. The technology is constantly improving as well as the analytical tools capable of identifying the potential genetic signatures in large data sets. Notwithstanding, critical evaluation of each individual finding is still paramount, last but not least due to the statistical pitfalls of potential type-2 errors.
Post mortem brain findings
An important target of investigations into gene expression is post mortem brain tissue, but one potential confounding factor is RNA degradation. To address this, Catts et al. (2005) quantified RNA over various post mortem intervals and observed good stability within the first 6 hours and acceptable RNA levels for up to 48 hours post mortem, but a significant decline thereafter. Hence, post mortem studies are nowadays generally required to cite RNA Integrity Number (RIN) as an index for potential RNA degradation. Brain pH is a significant factor that affects the quality of the RNA obtained from port mortem tissue (Bahn et al., 2001; Halim et al., 2008). Hence, post mortem intervals and tissue pH are generally reported and commonly matched when comparing patient and control data. Other commonly employed matching criteria are age, sex, co-morbidity and medication status. However, post mortem interval should not be viewed as a constant measure across cases as the environment after death may vary across cases with complex impact on levels of autolysis.
Most gene expression studies targeted grey matter while some studies investigated gene expression in immunohistochemically characterised single cells (Bahn et al., 2001). The latter approach is of particular interest since it is specific to individual cell types (Pietersen et al., 2009).
Common brain regions include: (1) the prefrontal cortex which is implicated in planning complex cognitive behaviours, decision-making and social cognition (Goldman-Rakic and Selemon, 1997; Pomarol et al., 2010; Yang and Raine, 2009); (2) the temporal lobes, including entorhinal cortex, which serves as the main interface between hippocampus and neocortex (Hargreaves et al., 2005); and (3) the hippocampus which forms a part of the limbic system and plays an important role in memory function and spatial information processing (Hargreaves et al., 2005; Harrison, 2004). All three brain regions have been implicated in schizophrenia (Pantazopoulos et al., 2010; Sivagnanasundaram et al., 2003) as well as interconnecting thalamic relay nuclei. For instance, Chu et al. (2009) screened the genome of laser-captured micro-dissected neurones of the primary thalamic relay nucleus of the dorsolateral prefrontal cortex and found IGF1-mTOR-, AKT-, RAS-, VEGF-, Wnt- and immune-related signalling, and eIF2- and proteasome-related genes were uniquely dysregulated in schizophrenia (n = 15) compared to patients with major depressive or bipolar disorder or healthy control subjects (n = 15 each).
Animal models suggest that down-regulation of oligodendrocyte transcripts in medial prefrontal cortex – thereby affecting myelinisation – is associated with impaired executive function that is commonly seen in patients with schizophrenia (Gregg et al., 2009). The prefrontal Brodmann’s area (BA) 9 was also the first human brain region to be investigated by microarray analysis by Mirnics et al. (2000). This study consisted of two groups of schizophrenia and control subjects with a total n of 11 for each matched pair that were matched for age, sex, post mortem interval, brain pH and brain storage time. Gene expression was determined using microarrays containing over 7000 features. The authors observed a significant decrease in expression of genes involved in prefrontal presynaptic function and confirmed using in situ hybridisation that the presynaptic function genes coding for N-ethylmaleimide-sensitive factor and synapsin II were consistently down-regulated. The group later also reported that the transcript-encoding regulator of G-protein signalling 4 (RGS4) was the most consistently and significantly decreased in the prefrontal cortex (BA9) in schizophrenia (n = 6) when compared to post mortem tissues of matched controls (n = 6) or patients diagnosed with major depressive disorder (n = 10) (Mirnics et al., 2001). Decreased RGS4 expression was also verified across three cortical areas of 10 subjects with schizophrenia with quantitative in situ hybridization. These findings suggest multiple small but synergistic effects on gene expression, linked to impaired nerve terminal function in schizophrenia.
Further support for this notion is derived from several findings such as the reduced expression of the synaptophysin gene in layer II stellate neurons in entorhinal cortex (Hemby et al., 2002). Synaptophysin is an integral membrane protein of small synaptic vesicles in the brain and endocrine cells and appears to be involved in organising membrane components and it therefore is easy to see how these changes could cause dysregulation of the synapse in schizophrenia. More recently, consistent expression changes were also identified in gene sets associated with synaptic vesicle recycling, transmitter release and cytoskeleton dynamics by Maycox et al. (2009) in the anterior prefrontal cortex tissue of BA10 when using high-density microarrays (30,000 features) to investigate two large schizophrenia cohorts.
A year after the first microarray study in schizophrenia, reduced expression of five oligodendrocyte and myelin-related genes were identified by Hakak et al. (2001) in prefrontal cortex (BA46) in a cohort of 12 schizophrenia patients versus matched controls. Altered expression of myelination-related genes have also been reported in other brain areas (Tkachev et al., 2007) such as CNP, MAG, OLIG2 and ErbB3 which were down-regulated throughout neocortex, hippocampus, caudate nucleus and putamen (Katsel et al., 2005). These findings are of particular relevance to schizophrenia, given that myelination is a key process of brain maturation.
In a well-controlled study of 14 schizophrenia brains, Arion et al. (2007) identified over-expression of immune chaperone function-related genes in the same cortical region. The authors used a study-specific DNA microarray platform with long oligonucleotides and multiple probes and replicates consisting of 1800 genes. The authors carefully matched their 14 patients on sex, age, post mortem interval, brain tissue pH, RNA integrity number and tissue storage time with an equal number of healthy control subjects. Another study indicated higher activity of histone de-actylase 1 in BA10 and 46 of prefrontal cortex suggestive of transcriptional repression due to increased lysine de-acetylisation in histone and non-histone proteins (Sharma et al., 2008). Kim et al. (2007) initially identified changed expression of 70 genes of which PCR confirmed a significant down-regulation of phospholipid scramblase 4 (a gene coding for a protein involved in apoptosis and blood coagulation) and empty spiracles homolog 2 transcription factor, which is involved in neurodevelopment. However, these expression changes were only observed in a cohort of suicide completers.
In a cross-study analysis of seven gene expression microarrays, Choi et al. (2008) identified 110 differentially expressed transcripts across various brain regions in 163 individuals with and without psychosis. In dorsolateral prefrontal cortex, PCR confirmed up-regulated metallothionein genes (which code for proteins with an affinity to heavy metals and glucocorticoids), while neuropeptide-related genes were found to be down-regulated. In an interesting study, Narayan et al. (2008) investigated duration of illness effects on gene expression in BA46 in three cohorts of matched pairs of schizophrenia and control subjects. The first cohort had a duration of illness of less than 4 years (n = 8 matched pairs), the second cohort between 7 and 18 years (n = 14 matched pairs) and the third of more than 28 years (n = 8 matched pairs). Early in the progression of schizophrenia, dysfunction to genes involved in gene transcription and RNA processing, vesicle-mediated transport functions, and metal ion binding was identified whilst long-term illness was associated with inflammation and immune responses amongst other various biological functions (Kasai et al., 2003). This suggested that gene expression changes as the disease progresses.
Tang et al. (2009) reported that normal aging was significantly linked to abnormalities in pathways related to synaptic function, cell-cycle/DNA damage and apoptosis. In contrast, aging in schizophrenia was significantly associated with fatty acid and steroid metabolism, but not with those functions associated with normal aging when investigating post mortem BA46 tissue from 29 schizophrenia and 30 healthy subjects aged from 19 to 81 years. Investigating the same brain region in 27 schizophrenia subjects and 27 matched healthy controls, Narayan et al. (2009) found differential expression of genes, particularly related to glycosphingolipid/sphingolipid metabolism and N- and O-linked glycan biosynthesis in schizophrenia. Expression decreases of seven genes associated with these pathways (i.e. UGT8, SGPP1, GALC, B4GALT6, SPTLC2, ASAH1 and GAL3ST1) were validated by quantitative PCR in schizophrenia subjects with short-term illness while only one of these genes (i.e. SPTLC2) was differentially expressed in chronic schizophrenia. Moreover, the expression of five of these genes was also significantly positively correlated with age in schizophrenia but not in control subjects. The authors concluded that a disruption of the sphingolipid metabolism in the early phase of illness could result in widespread downstream effects involving myelination and oligodendrocyte function, while the age-related effects may represent an adaptive response to disease progression or pharmacotherapy. The latter notion, however, was not supported by their data showing no correlation of gene expression levels with medication doses.
Torkamani et al. (2010) investigated post mortem BA9 and BA46 tissue from a large sample of 101 subjects and also confirmed differential age-related gene expression. In particular, the expression if genes related to developmental processes (i.e. neurite outgrowth, neuronal differentiation and dopamine-related cellular signalling) decreased with aging in normal subjects but not in schizophrenia subjects.
Temporal lobe neuropathology appears to be associated with some of the positive symptoms of schizophrenia (e.g. Kasai et al., 2003; Sivagnanasundaram et al., 2003), thus making the temporal cortex another prime target of molecular genetic research along with the prefrontal cortex. To this end, Hemby et al. (2002) reported decreased expression of genes associated with G-protein, glutamate and N-methyl-
More recently, Bowden et al. (2008) identified altered expression of genes involved in neurotransmission (linked to presynaptic functions), myelination and neurodevelopment when investigating post mortem brain tissue derived from the superior temporal gyrus of seven schizophrenia patients versus seven closely matched healthy control subjects. Interestingly, similarly altered expression profiles were also found in peripheral blood lymphocytes of schizophrenia patients (Bowden et al., 2006; see also below).
Subcortical brain structures that have been implicated in schizophrenia include the hippocampus and amygdala. Along with volume reduction and impaired glutamate neurotransmission, impaired function of the hippocampus has been linked to psychopathology and cognitive impairment in schizophrenia (e.g. Goff and Coyle, 2001; Heckers, 2001; Phillips et al., 2003). Chung et al. (2003) observed increased expression of chondrex (or YKL-40, which codes extracellular matrix glycoprotein involved in cell growth and migration), histamine-releasing factor, HERC2 and heat-shock 70. The histamine-releasing factor gene has been linked to negative symptoms, impaired learning and memory deficits in schizophrenia (Chung et al., 2003; Goff and Coyle, 2001) but was not confirmed by real-time RT-PCR and neither were HERC2 and heat-shock 70. Increased expression of chondrex, on the other hand, was confirmed, suggesting altered neuronal migration during neurodevelopment. In the amygdala, up-regulation of genes involved in presynaptic vesicle release (i.e. piccolo, RIMS2 and RIMS3) in the cytomatrix active zone as well as changes in genes involved in neurotransmission and myelination were observed in subjects with schizophrenia (Weidenhofer et al., 2006). In their follow-up study, Weidenhofer et al. (2009) showed that antipsychotic drug treatment was not likely to be the cause of the up-regulation of the genes involved in presynaptic release.
With hundreds of changes in gene expression being observed in different brain regions in schizophrenia, researchers more recently started to look for the mechanism(s) that might cause such widespread dysregulation of gene expression. One possibility that is gaining momentum is some evidence for increased global expression of microRNAs, which are involved in the regulation of translation of schizophrenia-related mRNA in superior temporal gyrus and dorsolateral prefrontal cortex in schizophrenia (Beveridge et al., 2010). Thus, one or several microRNA could be causing major changes in gene expression in schizophrenia.
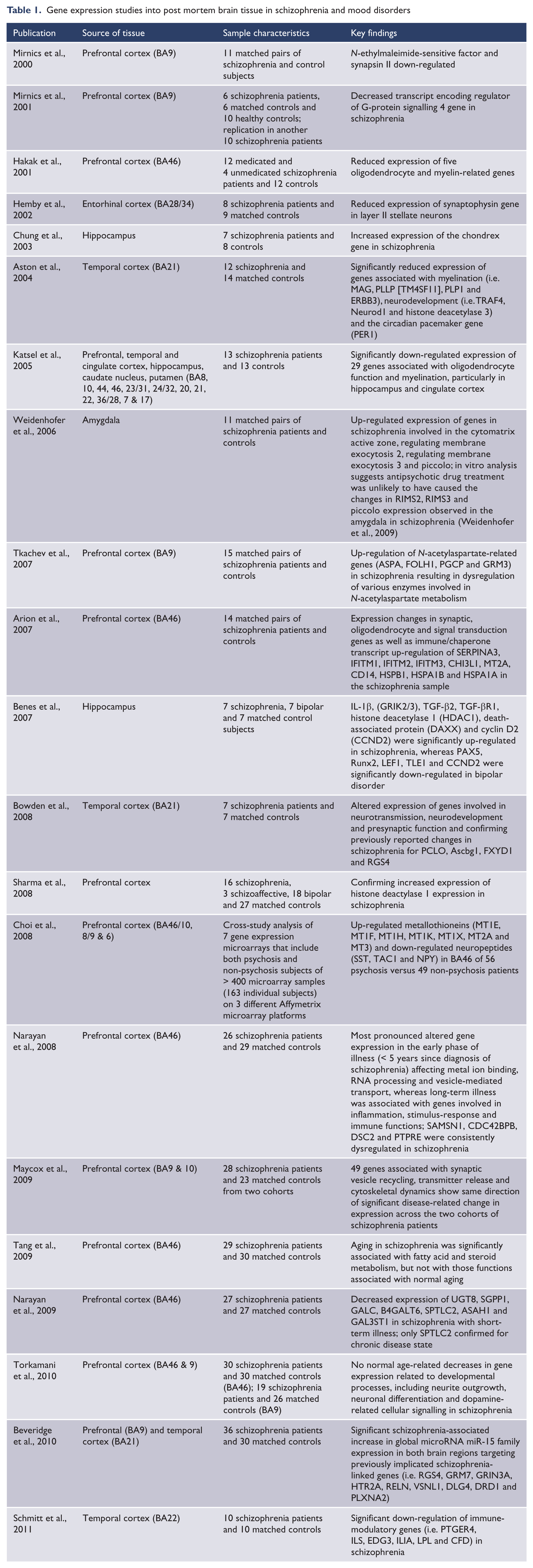
So far, the review has summarised the evidence for an array of altered gene expression in various brain regions implicated in schizophrenia (see Table 1 for summary). Taking these post mortem findings together, these genes are likely to affect synaptic processes such as vesicle release and neurotransmitter signalling (reviewed by Eastwood and Harrison, 2001) as well as neurodevelopmental processes such as myelination and neural migration. These are all very plausible changes that could lead to schizophrenia. However, post mortem brain research is limited by the availability of suitable tissue – thus, usually resulting in small sample sizes – and it also relies on the quality and accuracy of existing medical records of deceased tissue donors. The use of peripheral tissues and cell samples for sources of RNA like blood has been investigated as an alternative to post mortem brain tissue. The choice of peripheral tissue may address some of these limitations but also raises the question of whether non-neural cells can inform about the brain (Matigian et al., 2008).
Gene expression studies into post mortem brain tissue in schizophrenia and mood disorders
Gene expression findings from peripheral tissues
In contrast to post mortem brain tissue, peripheral blood mononuclear cells (PBMC) can be easily collected from large cohorts for gene expression studies. It also has the advantage that patients can be phenotyped and followed-up longitudinally. PBMC gene expression studies have targeted putative risk factors for schizophrenia as well as potential biomarkers of treatment response and prognosis (Bartolomucci et al., 2010; Huang et al., 2008; Kennedy, 1996; Tsuang et al., 2005; Yao et al., 2008). There have also been two types of studies conducted, those using fresh PBMCs and those that have used transformed PBMCs into immortalised lymphoblastoid cell lines (LCLs). These cell transformations, however, may introduce a confound on gene expression profiles, thus making it potentially difficult to separate genuine disease effects from disease by cell culture interactions.
In 2006, Vawter et al. (2006) reported gene expression profiles from LCLs obtained from a multiplex schizophrenia pedigree containing five individuals with schizophrenia and nine unaffected family members. A total of nine genes were altered by array analysis, but only in two of those the neuropeptide Y receptor Y1 gene and the human guanine nucleotide-binding regulatory protein Go-alpha were confirmed to be significantly decreased by RT-PCR.
Following on from this were three studies of fresh PBMCs. The first used PBMCs from 28 controls, 30 individuals with schizophrenia and 16 with bipolar disorder (Tsuang et al., 2005). This study showed that gene expression profiling of RNA from PBMCs could distinguish between schizophrenia and bipolar disorder. This was supported by the second study using PBMCs from sib-pairs with schizophrenia (n = 66 siblings) and bipolar disorders (n = 10 siblings) that also detected more than 2000 genes with altered expression in schizophrenia. Interestingly, some of these genes were from functional groups involved in processes such as neurotransmission and presynaptic function (Middleton et al., 2005). In the third study, Bowden et al. (2006) observed altered expression of 18 brain-related genes in PBMC derived from 14 drug-treated schizophrenia patients versus matched healthy control subjects. These studies suggested that gene expression changes in PBMCs might reflect changes in the brain. Indeed, Sullivan et al. (2006) compared gene expression profiles from PBMCs and various brain tissues, including the prefrontal cortex, and found significant overlap in the expression of genes in the blood and the brain which, however, does not mean that these peripheral gene expression changes also translate into potential effects on brain function.
The use of fresh PBMCs still does not mitigate antipsychotic drug treatment. In order to do this, studies need to be conducted in treatment-naïve participants. To this end, when comparing PBMC gene expression of 13 drug treatment-naïve schizophrenia patients with matched healthy control volunteers, Zvara et al. (2005) found PCR-confirmed increased expression of the inwardly rectifying potassium channel (Kir2.3) and dopamine-3 receptor genes. Craddock et al. (2007) also showed prominent transcript changes in genes involved with cell-cycle machinery, intracellular signalling, oxidative stress and metabolism in schizophrenia patients (compared to controls) in a sample of six schizophrenia patients versus six control subjects by using human whole-genome microarray on freshly isolated T-cell-derived RNA. Subsequent chromosomal location analysis of altered genes showed susceptibility clusters at 1p36, 1q42 and 6p22 which had been previously implicated with schizophrenia (Sivagnanasundaram et al., 2003).
Another line of research has focused on undifferentiated cell lines or blast cells derived from olfactory neuroepithelium showing dysregulated cell-cycle and phosphatidylinositol signalling in schizophrenia and bipolar I disorder, respectively (McCurdy et al., 2006). More recently, Glatt et al. (2009) investigated alternatively spliced genes as potential biomarkers in peripheral blood mononuclear cells of 13 schizophrenia, nine bipolar and eight healthy control subjects. Their preliminary finding suggests significant interactions between diagnostic group and exon identity, with 33 genes showing differential splicing patterns between schizophrenia and bipolar disorder. The group also reported dysregulated ubiquitin proteasome pathways for psychosis and bipolar diagnostic groups in two independent samples (Bousman et al., 2010).
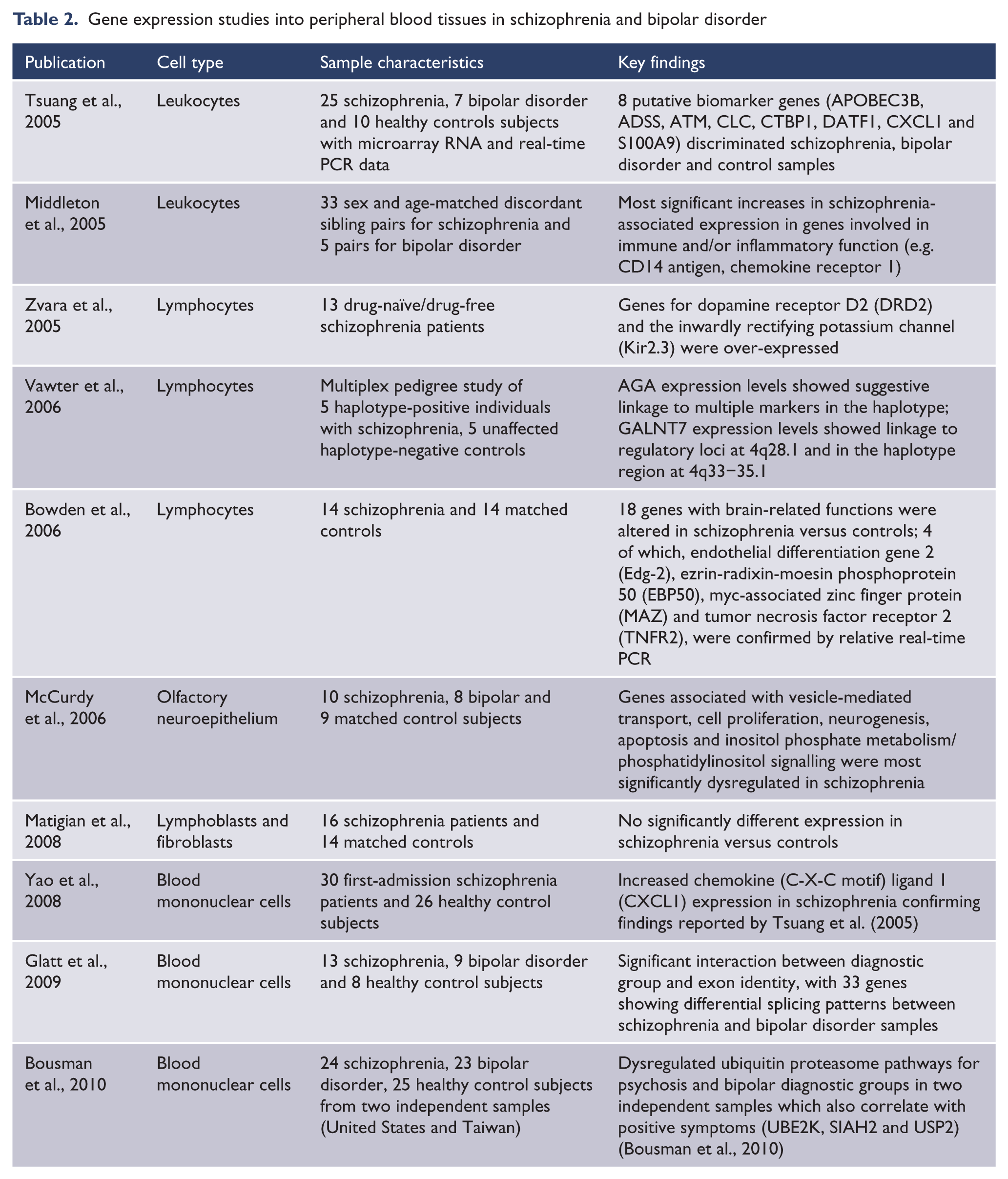
This line of research, however, stills lacks consistency and has not produced exclusive biomarkers or other signatures that may assist with diagnosis (see Table 2 for summary). This is mostly due to the heterogeneous nature of schizophrenia and the small sample sizes, which are prone to false-positive results. The few findings reported here also show little overlap with post mortem reports, making extrapolations to impaired brain functions difficult. Nevertheless, gene expression studies of peripheral tissues gain momentum in schizophrenia research but are very much dependent on systematic research of transient (e.g. medication effects) and more stable illness effects (i.e. associated with susceptibility genes). Importantly, the in vivo nature of this type of research allows for longitudinal tissue collection in parallel with observing the clinical phenotype.
Gene expression studies into peripheral blood tissues in schizophrenia and bipolar disorder
Animal models research
With the advances of genetically modified animals, the effects of genome alterations on gene expression have become an important area of schizophrenia research. For instance, 22q11.2 deletion syndrome probably accounts for approximately 1% of all schizophrenia patients (Bassett and Chow, 2008) whereas up to 35% of the individuals with the deletion are likely to develop schizophrenia.
Jurata et al. (2006) and Sivagnanasundaram et al. (2007) investigated a mouse model by deleting 1 million base pairs in a region of chromosome 16. The resulting Df1/+ mouse model carries a hemizygous deletion in a region which corresponds to the human 22q11.2 region. The heterozygous (Df1/+) mice display deficits in prepulse inhibition, learning and memory, consistent with the schizophrenia phenotype (Blundell et al., 2010).
Sivagnanasundaram et al. (2007) identified 159 differentially expressed genes in heterozygous (Df1/+) mice, with 12 genes mapping to the deleted region and expressed in hippocampus. Moreover, 15 genes involve signal transduction, synaptic plasticity, neuronal differentiation, microtubule assembly and a ubiquitin pathway relevant to hippocampus-mediated function as confirmed by PCR. Earlier, Jurata et al. (2006) reported significantly reduced expression of COMT and PRODH genes in hippocampal dentate granule neurons.
Rodents with ventral hippocampus lesions have also been introduced as an animal model for schizophrenia (Lipska, 2004). Wong et al. (2005) showed that treatment with the antipsychotic haloperidol normalises the expression of those genes where the expression has been altered by the lesion. Intrapartum viral infection has also been linked to a higher prevalence of schizophrenia (Wright et al., 1995). When infecting mice with influenza virus at day 18 of pregnancy, altered expression of immunoglobulin domain (Ig), secreted semaphoring 3A (Sema3a), transferrin receptor 2 (Trfr2) and Vldlr were identified by microarray and confirmed with PCR in hippocampus, prefrontal cortex and cerebellum (Fatemi et al., 2008, 2009).
Isolation rearing has also been introduced to study the effects of an environmental insult on neurodevelopment. Genes related to GABA neurotransmission and synapse structure (Gabra4, Nsf, Syn2 and Dlgh1) were found to be over-expressed in prefrontal cortex of affected rat pups along with reduced extracellular glutamate levels and under-expression of transcripts related to glutamatergic transmission and synapse integrity in the adult animals (Murphy et al., 2010).
Genetically modified animals have become an important tool when investigating the potential biological mechanisms of the pathogenesis of schizophrenia. Studying altered gene expression in these animal models is another logical step when considering the complexity of effects which are arising from single gene knock-outs. However, owing to the complex nature of the genetics of schizophrenia, single gene knock-out animal models are highly unlikely to be representative of the schizophrenia syndrome. Thus, to date, any animal model is limited to certain aspects of the disorder – such as individual aspects of micro or macro neuropathology or pathophysiology – with the complete clinical phenotype remaining an exclusive human phenomenon. Moreover, the effects of knocking out individual genes can also considerably differ between strains of mice; for instance, being potentially lethal in one strain and not having any discernable effect in another.
Moreover, gene expression changes have been reported across diagnostic categories. For instance, Kanazawa et al. (2008) reported up-regulated SELENBP1 in post mortem dorsolateral prefrontal cortex of patients diagnosed with schizophrenia and bipolar disorder whereby the expression of SELENBP1 was significantly correlated with the presence of psychosis across diagnoses. Hence, animal modelling may also need to take into account that syndrome-based diagnostic categories potentially represent a variable phenotype of a common pathological mechanism.
Conclusion
Microarray gene expression studies into schizophrenia are still inconsistent in their findings. While some findings have linked altered gene expression to presynaptic function, signalling, myelination, neural migration, cellular immune mechanisms and response to oxidative stress, the majority of results are difficult to interpret due to small sample sizes (i.e. potential type-2 errors), confounding factors (i.e. medication effects) or lack of plausible neurobiological theory. By complementing structural genetic research into the origin of schizophrenia, microarray gene expression studies are likely to play an important role in the future when investigating gene/gene and gene/environment interactions. For instance, longitudinal research into PBMC or other in vivo tissue holds the promise of identifying gene expression profiles that are associated with phenotype changes (e.g. acute versus chronic illness; prodromal versus established illness, etc.) and environmental factors (e.g. on and off medication treatment; contributing effects of substance abuse; etc.). Experimental animal models, on the other hand, are particularly useful when studying changes of gene expression in genetically modified animals, including the effects of environmental stressors on brain development. Post mortem brain research, however, relies on a scarce resource which should not be wasted when alternative methods such as in vivo tissue and animal models are available. Nevertheless, post mortem human tissue research will remain an important prerequisite to validate findings of in vivo and animal model research.
Footnotes
Funding
NK was supported by the World Health Organization, the Schizophrenia Research Institute and the University of Newcastle, and received infrastructure support from the Australian Schizophrenia Research Bank, the Hunter Medical Research Institute and New South Wales Health.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.