
Abstract
Background
Symmetric dimethylarginine (SDMA) and asymmetric dimethylarginine (ADMA) are naturally occurring amino acids classed as uraemic toxins by the European Uremic Toxins Work Group. SDMA is principally excreted through the kidneys and is a well-known renal function marker, and ADMA is a potent inhibitor of nitric oxide production. Here, we describe the development of a rapid and sensitive liquid chromatography tandem mass spectrometry method for simultaneous measurement of SDMA, ADMA and creatinine.
Method
Serum samples were prepared by protein precipitation and dilution with acetonitrile prior to injection onto a Waters TQS-Micro. SDMA, ADMA, creatinine and their corresponding internal standard transitions were detected using multiple reaction monitoring after separation with a hydrophilic interaction liquid chromatography analytical column. Sample stability and intra-individual variation studies were also assessed following ethical approval.
Results
The retention time for creatinine was 0.43, SDMA 1.10 and ADMA 1.14 min. Mean recovery for creatinine was 103%, SDMA was 100% and ADMA was 103%; matrix effects were minimal (<6%). Lower limit of quantitation for creatinine and SDMA/ADMA was 17.5 µmol/L and 0.1 µmol/L, respectively. Analytical imprecision showed a coefficient of variation <10% for all analytes across the working range of the assays. Intra-individual variation for creatinine was 4.7%, SDMA 7.5% and ADMA 7.6%.
Discussion
We have developed a rugged assay for measurement of SDMA, ADMA and creatinine by LC-MS/MS suitable for routine use. It is easy to perform owing to its simplicity and reproducibility. The stability of SDMA and ADMA pre- and post-centrifugation allows for their routine use without any special sample handling requirements.
Introduction
Through post-translational modifications of arginine, symmetric dimethylarginine (SDMA) and asymmetric dimethylarginine (ADMA) are naturally occurring amino acids generated intracellularly. SDMA and ADMA are classed as uremic toxins by the European Uremic Toxins Work Group (EUTox) and are both independent risk markers for all-cause mortality.1,2 In addition, they are strongly linked to cardiovascular disease, immune dysfunction and negative disease outcomes in critically ill patients. Through inhibition of nitric oxide synthase, ADMA is the most potent endogenous inhibitor of nitric oxide production 3 and is therefore implicated in endothelial dysfunction, atherosclerosis and consequently cardiovascular disease.
ADMA is primarily cleared by enzymatic degradation in the liver and kidney by 2 forms of dimethylarginine dimethylaminohydrolase (DDAH). The majority is metabolised to monomethyl amine and dimethylamine with the remaining 10%–20% of ADMA being renally cleared thus ADMA does not serve as a useful renal function marker.4,5 SDMA on the other hand is principally excreted by the kidneys, hence it’s potential use as a renal function marker. Previous studies have shown it to be a better marker of renal function than CKD-EPI and MDRD derived eGFR. 6
The aim of this project was to develop and validate a combined LC-MS/MS method for the measurement of SDMA, ADMA and creatinine in serum samples. The addition of creatinine will allow direct comparisons to be undertaken in any forthcoming clinical studies. Current analytical methods utilise derivitisation with solid phase extraction (SPE) separation, 7 use of expensive chiral columns or long chromatographic separation times which may hinder their introduction into a routine clinical laboratory setting. 8 Despite SDMA and ADMA being isobaric, chromatographic separation is not necessary due to the unique transitions present, therefore allowing fast chromatographic methods to be utilised.
In addition to the development of the assay we will undertake an intra-individual variation (IIV) study using healthy laboratory volunteers. Application for ethics has been undertaken (IRAS 273368) with a favourable REC review. A previous IIV study has been published using HPLC with a fluorimetric detection, 9 but to the author's best knowledge, there are no IIV studies for SDMA/ADMA using LC-MS/MS as the analytical technique.
Materials and methods
Intra-individual variation
Surplus serum samples were anonymised and used for method validation and sample comparison purposes. The IIV study enrolled 28 healthy laboratory volunteers (12 males and 16 females) who completed an enrolment questionnaire after reviewing the study protocol and supporting information. Fasting venous samples were taken once a week for 10 weeks and collected into SST™ II Advance Plus blood collection tubes (Becton Dickinson, Wokingham, UK). Samples were aliquoted and serum stored at −80°C prior to triplicate analysis on the same analytical run. The IIV coefficient of variation as a percentage was calculated using the following formula where A = analytical variation and T = total variation.
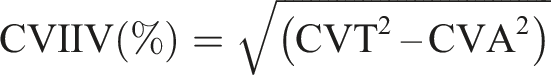
Sample stability
To assess the impact of a delay in centrifugation, an additional 8 serum tubes were collected from 12 study participants. After collection, samples were allowed to clot for 3 min and subsequently centrifuged at different time points (2, 4, 8, 12, 24 and 48 h) to emulate a delay in sample receipt in the laboratory. Immediately after centrifugation at each time point, 1 mL of sample was aliquoted and frozen at −80°C prior to triplicate analysis on the same analytical run. In addition, to better understand the impact of leaving serum on the gel, samples that had been centrifuged were stored at 4°C and aliquoted at different time points (8, 24, 48, 72 and 96 h) prior to analysis. Samples were compared to the baseline result and a percentage difference calculated.
For each analyte, the reference change value (RCV) was calculated using the following equation where A = analytical variation and B = biological variation:
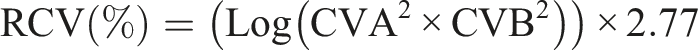
Calculation of RCV thus provides acceptability criteria for the stability studies undertaken as part of this study
10
SDMA RCV = 18.0% ADMA RCV = 23.0% Creatinine RCV = 13.8%
Sample preparation
Approximately 10 mg of powdered SDMA, ADMA and creatinine pharmaceutical secondary standard (Sigma, Dorset, UK) were gravimetrically added to 50% methanol (HPLC-MS grade, Honeywell, Seelze, Germany) to make quality control (QC) and calibrator stock solutions. To make working standards, these were subsequently diluted in phosphate buffered saline (PBS) (Sigma, Dorset, UK) to produce concentrations ranging from 0 to 3 µmol/L for SDMA/ADMA and 0 – 400 µmol/L for creatinine. QC material was made in the same way from a different intermediate stock solution. Internal standards (d6-SDMA, d6-ADMA and d3-creatinine (TRC, Toronto, Canada)) were combined in acetonitrile to a working concentration equivalent to 1 µmol/L for SDMA/ADMA and a creatinine concentration of 100 µmol/L. Storage for standards, QCs and combined internal standard was at −20°C prior to use. Extraction buffer was made prior to sample preparation and was stable for 1 week at room temperature (0.5% formic acid and 80 mmol/L ammonium formate (Sigma, Dorset, UK) in deionised water).
Sample preparation procedure was based on a modified published method by Lobenhoffer et al. 11 Samples, standards and QCs (10 µL) were added to a 2 mL 96-well plate (Porvair, Leatherhead, UK) using a positive displacement pipette for increased accuracy. To each well, extraction buffer (60 µL) was added, followed by internal standard (20 µL) and 600 µL of HPLC-MS grade acetonitrile (Honeywell, Seelze, Germany). The plate was sealed and vortexed (1 min at 1800 rpm) prior to centrifugation (5 min at 880 g).
Chromatography
Mobile phases consisted of 40 mmol/L ammonium formate 0.4% (v/v) formic acid (BDH, Bristol, UK) in deionised water (A) and acetonitrile (B). Sample extract (2 µL) was injected onto a 2.7 µm CORTECS hydrophilic interaction liquid chromatography (HILIC) 2.1 × 30 mm analytical column coupled with an in-line filter (both Waters, Manchester, UK) with a flow rate of 0.5 mL/min. This differed from the original published method by using a different column and without using TFA in the mobile phase. Starting conditions were 15% A which was held for 0.2 min before increasing linearly to 35% A over 0.1 min, this was held for 0.7 min prior to returning to starting conditions. Time per injection was 1.5 min in total.
Mass spectrometry
A Waters XEVO® TQS-Micro tandem mass spectrometer controlled by MassLynx 4.1 software was used in electrospray positive mode. Capillary voltage was 0.4 kV and source temperature was 150°C. The desolvation temperature and gas flow were 500°C and 1000 L/h, respectively. The quantifier transitions monitored for SDMA were m/z 203 > 172, ADMA m/z 203 > 46 and creatinine 114 > 44. These transitions were detected in multiple reaction monitoring (MRM) mode with a dwell time of 31 Ms. TargetLynx was used to process data, each analyte was quantified using the ratio of analyte peak height to internal standard peak height against concentration (µmol/L). A 1/X weighting and linear least squares regression model was used.
Validation
The assay was validated for use in the laboratory following published acceptance criteria. 12
Accuracy
In the absence of matrix-matched certified reference material, gravimetrically prepared PBS-based QC material was used to assess accuracy of SDMA/ADMA. Quality control (QC) samples were analysed over 10 consecutive batches, a deviance from the target concentration of <15% was deemed acceptable. 12 For creatinine, UKNEQAS external quality assurance (EQA) samples (n = 8) were analysed and compared to the enzymatic target (method principle mean used), comprising of >400 participants with 5 different groups.
Recovery
SDMA/ADMA (0, 5, 1 and 2 µmol/L) and creatinine (0, 75, 150 and 300 µmol/L) prepared in 50% methanol were spiked into six different serum samples. Analyte recovery was calculated as a percentage with acceptability criteria of 100 ± 20%; endogenous SDMA/ADMA and creatinine were factored in when performing calculations.
Matrix effects
Both qualitative and quantitative experiments were carried out to investigate ion suppression or enhancement. For the qualitative approach, six different serum samples were extracted with the internal standard omitted. The samples were injected with the addition of a constant post-column infusion of internal standard. 13
A documented procedure was followed for quantitative assessment of matrix effects. 14 A PBS blank sample matrix and 6 serum samples were extracted as per the experimental protocol. After extraction, SDMA, ADMA and creatinine were added at the same concentration as the recovery spikes. Matrix effects were calculated as a percentage from the peak height relative to the PBS blank used after subtracting the endogenous analytes present.
Imprecision
Pooled serum and PBS-based QC samples spiked with known concentrations of SDMA, ADMA and creatinine were used to assess analytical imprecision. Analysis of each level of QC 10 times within the same sample run was done to assess intra-assay imprecision (repeatability). To calculate inter-assay (intermediate) imprecision, the same lot of QC material was analysed on 10 different analytical runs on different days. A coefficient of variation (CV) up to 15% was considered acceptable.
Linearity
A calibration curve containing SDMA/ADMA concentrations of 20 µmol/L and a creatinine concentration of 3000 µmol/L was made in PBS. Serial dilutions were carried out up to a factor of 32 to assess linearity above the top standard. Dilutional linearity was assessed by serially diluting samples (n = 4) with concentrations within the measuring range for each analyte. Serial dilutions were carried out up to a factor of 16 to confirm if a linear relationship exists between concentration and response. For all dilutions, we considered a deviation from the original concentration of <10% acceptable.
Carryover
To assess carryover, a PBS blank was analysed after the top calibration standard. If carryover was <20% of the lower limit of quantification, then this was deemed acceptable.
Lower limit of quantification (LLOQ)
A PBS sample with a SDMA/ADMA concentration of 0.1 µmol/L and a creatinine concentration of 18.75 µmol/L was analysed 10 times. To confirm the LLOQ in matrix samples, a serum sample with similarly low concentration was analysed 10 times within the same analytical run. To confirm inter-assay imprecision at the LLOQ, the PBS and serum-based samples were analysed on 10 different analytical runs on different days. If the bias <20% in the PBS sample and the imprecision was <20% in the both the PBS and matrix samples, the LLOQ was deemed acceptable. 12
Interfering substances
Serum samples (x3) with a range of SDMA, ADMA and creatinine concentrations were each spiked with haemoglobin (500 mg/dL), bilirubin (500 µmol/L) and intralipid (500 mg/dL) to assess any impact of common interferents. The native samples (no interference) were analysed at the same time, a difference ±10% was deemed acceptable.
Stability post-extraction
Patient samples (n = 15) were prepared and analysed. Immediately, afterwards the plate was resealed and stored at 4°C for 24 h to emulate analytical failure overnight. After this time, the plate was centrifuged and re-injected with any difference calculated as a percentage. A target of ±10% was deemed acceptable.
Comparison
Comparison of samples was undertaken to further assess accuracy. Serum samples were extracted and subsequently analysed using a UKAS accredited Roche enzymatic creatinine assay (linear range 5–2700 µmol/L). For comparison of SDMA and ADMA, commercially available ELISA kits (Immundiagnostik AG, Bensheim, Germany) were used. Samples were prepared in duplicate following the manufacturer’s instructions.
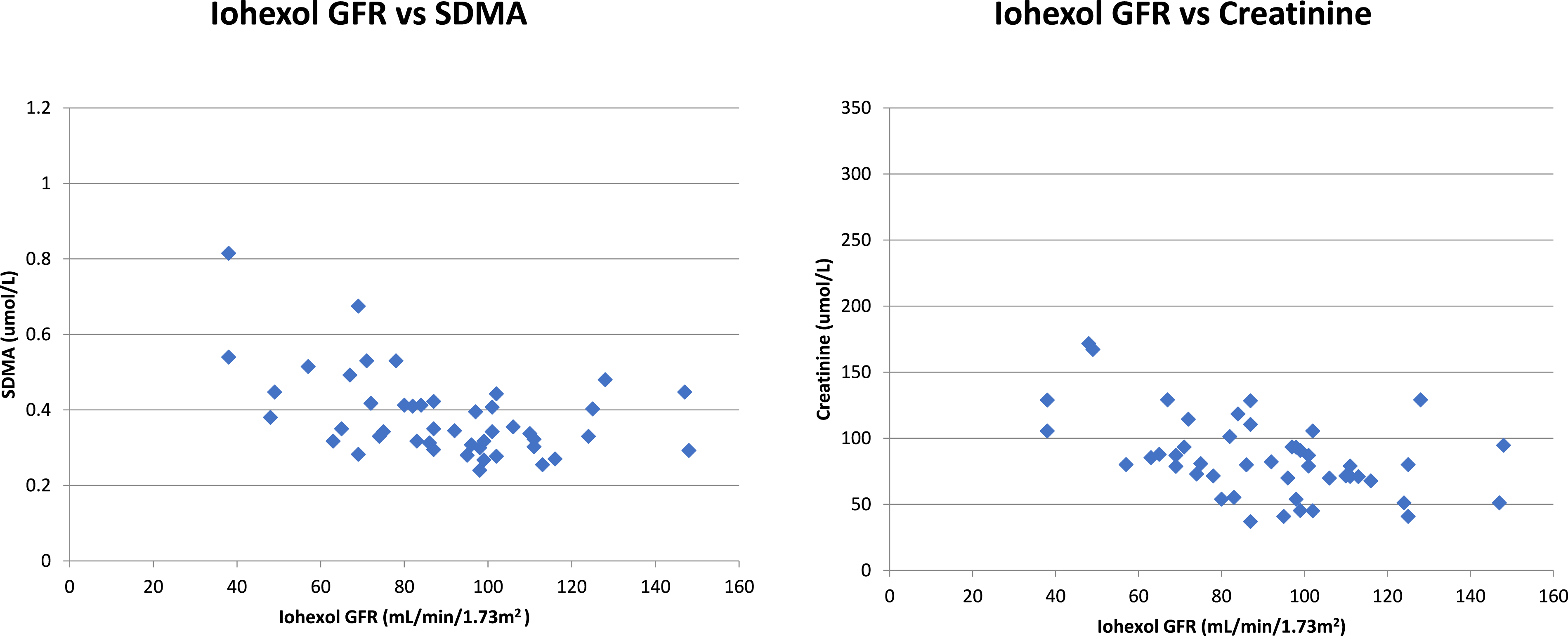
A fully validated LC-MS/MS assay was used to measure iohexol concentration in 48 patients who had undergone a measured GFR procedure based on plasma clearance of iohexol. The measured GFR was generated using well-established calculations and then compared to creatinine and SDMA concentration in the same patient samples to assess correlation. 15
Statistical analysis
Analyse-It Software (Analyse-It Software Ltd, Leeds, UK) was used for all data comparisons and analysis. Passing-Bablok regression analysis and Pearson correlation coefficients were used for method comparisons.
Results
Creatinine retention time was creatinine was 0.43, SDMA 1.10 and ADMA 1.14 min (Figure 1). Ion suppression through qualitative assessment was minimal; quantitative ion suppression (mean) was <6% for SDMA, ADMA and creatinine. Mean (range) recovery for SDMA was 100% (94%–109%), ADMA was 103% (94%–111%) and creatinine was 103% (99%–107%): all within acceptable limits. Chromatograms showing SDMA, ADMA and creatinine in a serum sample at the respective LLOQ.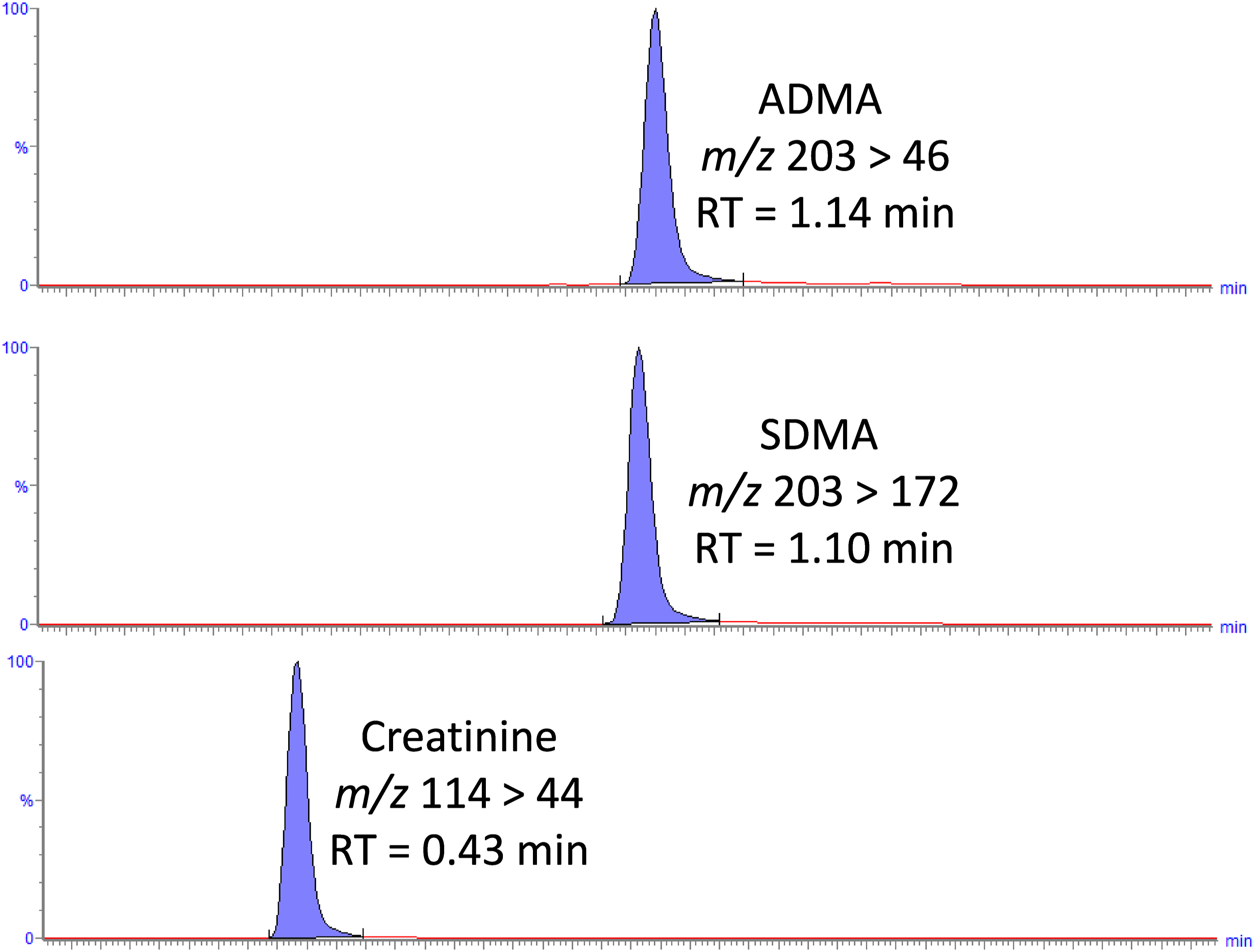
Calibration curves were linear showing inter-batch reproducibility, the R2 values were ≥0.999 for SDMA, ADMA and creatinine, (n = 20). Dilutional linearity experiments confirmed analyte concentration: response was linear, and that PBS was a suitable diluent for any samples with SDMA/ADMA concentrations up to 20 µmol/L and creatinine up to 2500 µmol/L.
The inter- and intra-batch imprecision and bias for PBS-based QC material were both <10% for SDMA, ADMA and creatinine; for serum samples, the CV remained <10% for both parameters (data not shown). The LLOQ was 0.1 µmol/L for both SDMA and ADMA, and 17.5 µmol/L for creatinine. The signal:noise ratio (s/n) for each analyte at the LLOQ was >10. For matrix and non-matrix samples, the imprecision (%CV) and bias were <10%. Absolute carryover was below the acceptable criteria of 0.02 µmol/L (SDMA/ADMA) and 3.5 µmol/L for creatinine (20% of the LLOQ). Comparison of creatinine EQA samples (n = 8) showed the LC-MS/MS assay to have an average positive bias of 1.5% with some scatter (range −3.6% to 8.1%).
For the assessment of post extraction stability, each analyte had a mean difference of <6% when stored at 4°C for 24 h.
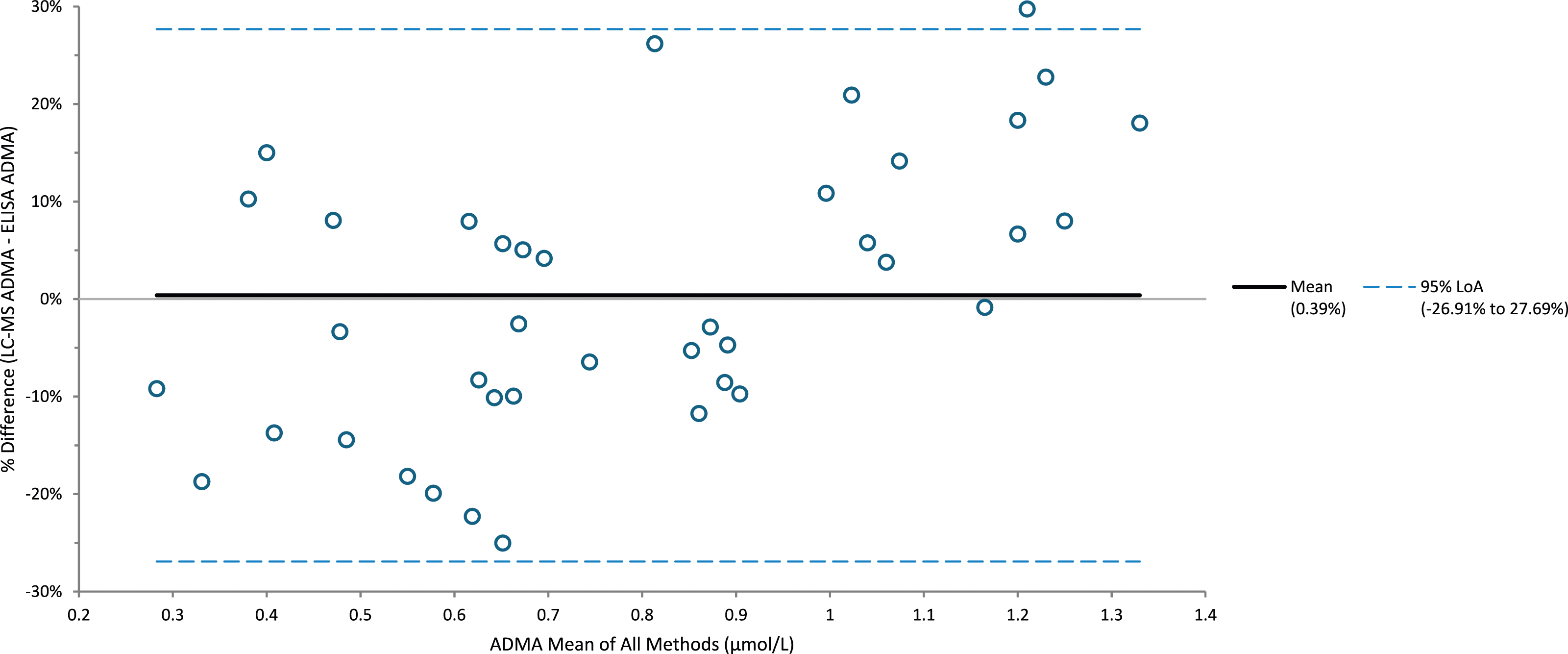
Comparison of SDMA samples (n = 35) with a commercial ELISA kit had an R2 of 0.976; the LC-MS/MS assay had a negative bias of −7.6% (Figure 2). Comparison of ADMA samples (n = 40) with a commercial ELISA kit had an R2 of 0.891; the LC-MS/MS assay had a positive bias of 0.4% (Figure 3). Comparison of creatinine samples (n = 40) with a Roche enzymatic method had an R2 of 0.999; the LC-MS/MS assay had a positive bias of 5.3%. Iohexol mGFR versus creatinine and SDMA highlights a similar pattern of results with similar correlation throughout the range of samples measured (Figure 4). Percentage difference of SDMA when analysed using LC-MS/MS and ELISA (n = 35). Percentage difference of ADMA when analysed using LC-MS/MS and ELISA (n = 40). Iohexol MGFR vs SDMA/creatinine, both exhibit a clear correlation further highlighting the utility of SDMA as a renal function marker.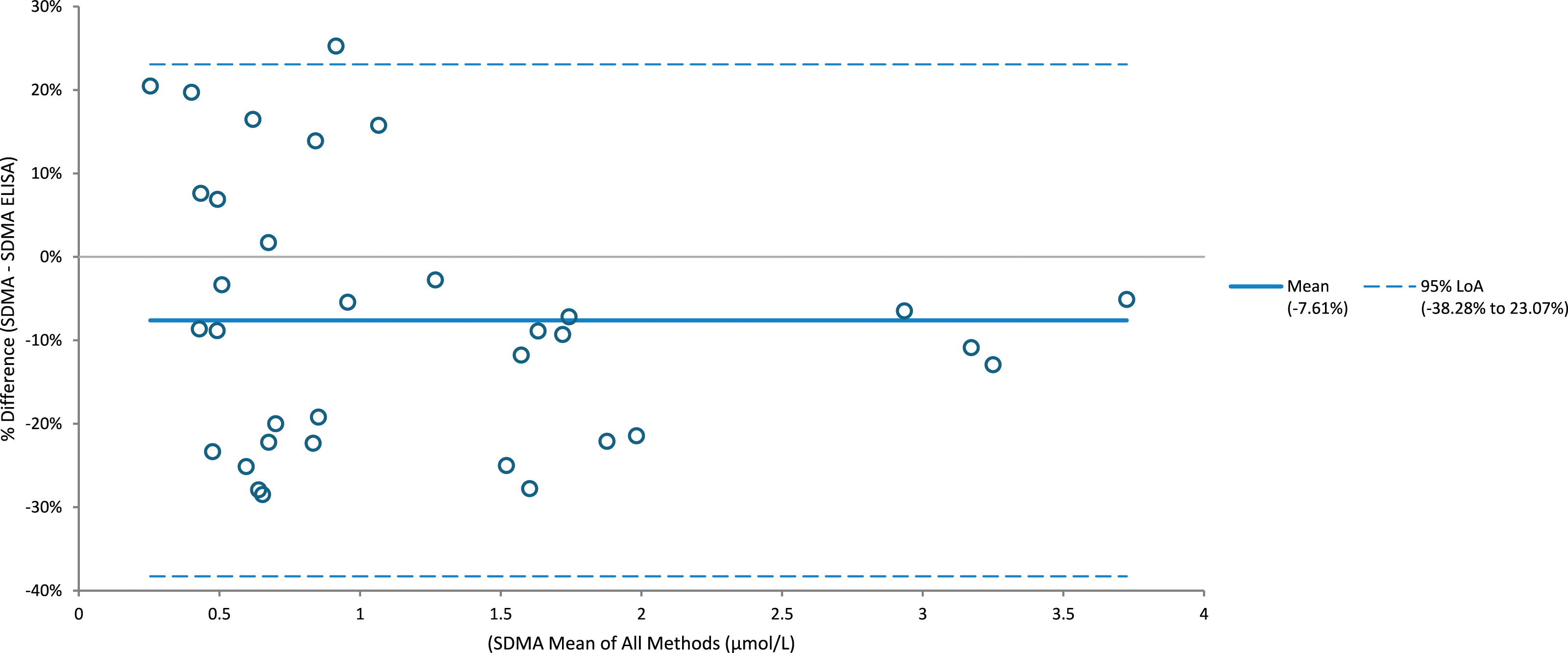
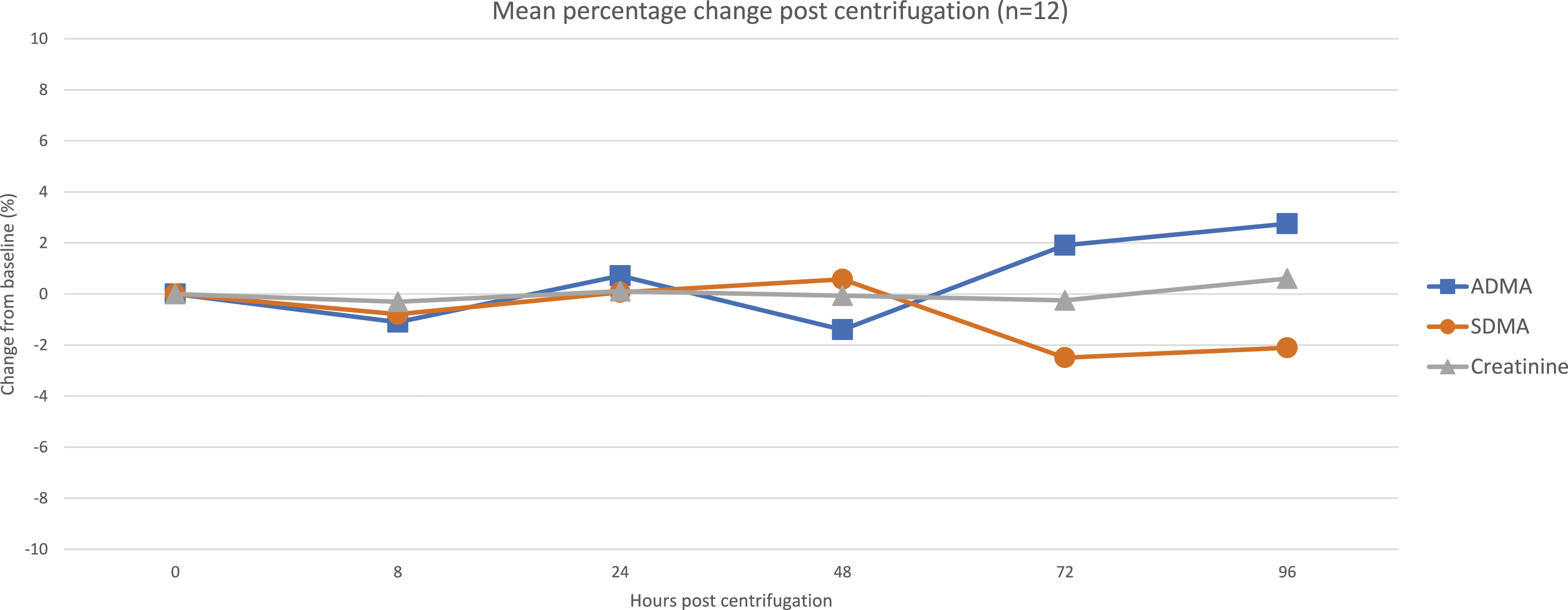
The intra-individual variation (IIV) for SDMA was 5.2%, ADMA 4.9% and creatinine 3.7%. Assessment of pre-centrifugation stability (Figure 5) showed large increases in SDMA and ADMA (15% and 50%, respectively, at 72 h), whilst creatinine remained unchanged. Changes after 96 h post-centrifugation (Figure 6) were much less pronounced for SDMA and ADMA (2% at 96 h), and once again, creatinine remained unchanged. Pre-centrifugation stability data of SDMA, ADMA and creatinine. The mean value for each measurand has been calculated and represented as a percentage difference from the baseline value. Post-centrifugation stability data of SDMA, ADMA and creatinine. The mean value for each measurand has been calculated and represented as a percentage difference from the baseline value.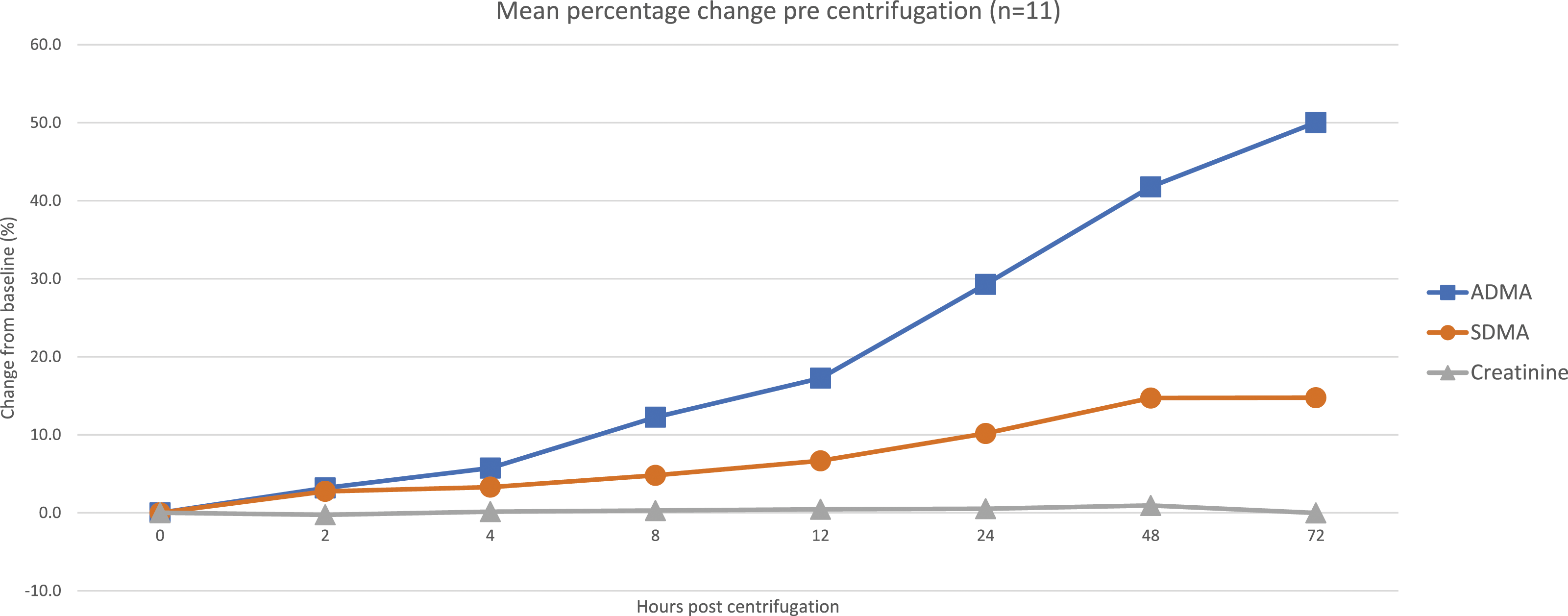
Discussion
HILIC was required to ensure adequate separation of the analytes due to their polar nature. Traditionally, re-equilibration of HILIC columns is slow; however, the use of a solid-core analytical column allows for faster re-equilibration times and consequent higher throughput. In addition, the sample preparation was simple, quick and can be undertaken using common laboratory reagents and equipment. This method is faster and simpler to perform than most other published methods, thus allowing for rapid sample turnaround to aid with clinical decision making.
Reproducible separation and peak shape were obtained with the chromatographic conditions described, remaining of high quality over the course of the validation and subsequent sample analysis. The described method exceeded validation requirements and has proven to be robust and reliable. Ion exchange chromatography was also investigated but there appeared to be interference present which caused a positive bias when results were compared with this HILIC method; hence, we did not proceed to the validation stage.
The assessment of IIV for SDMA and ADMA in 28 laboratory staff agreed with a previous study where the calculated IIV was 5.8% and 7.4%, respectively. 9 EFLM biological variation database stated the median creatinine IIV to be 4.5% (CI = 4.2%–5.7%), 16 in keeping with our calculated CV of 4.7%. Although the SDMA and ADMA IIV are greater than creatinine, there is still tight homoeostatic control in health – a requirement for a precise clinical assay.
A previously published study showed that serum and EDTA/lithium heparin plasma gave SDMA/ADMA results within 2% of one another proving anticoagulants have a minimal effect. 17 Owing to accessibility of serum samples in a routine clinical laboratory, we undertook assessment of stability prior to centrifugation. This showed that both SDMA and ADMA increase significantly – to the author’s knowledge, this has not been carried out previously, this discovery will hopefully inform other groups so their protocols can be amended accordingly. It is recommended that samples are centrifuged within 12 h of collection to ensure the change is within the calculated reference change values (SDMA = 18%, ADMA = 23% and creatinine = 13.8%). To enable sampling with plasma samples, this study needs to be repeated using EDTA and lithium heparin to assess the effect of delayed separation using alternative sample preservatives.
Further stability studies showed that extended (96 h) storage of centrifuged serum samples left on the gel had a minimal effect (<3%) on SDMA, ADMA and creatinine results. Proving that retrospective analysis of centrifuged stored samples would be feasible.
Comparison of creatinine in a range of surplus serum samples was acceptable proving the assay to be reliable and linear, and analysis of 8 EQA samples proved the method’s accuracy. Comparison of SDMA showed good correlation with a small positive bias. Interestingly ADMA correlation was not that good; low results had a positive bias whereas higher results had a negative bias. This could be due to differences in calibration used or problems with the dynamic range of the ELISA kit calibration curve, and previous papers have also shown discrepancies with ADMA being overestimated when analysed using ELISA.18,19
To the author’s knowledge, there is no certified reference material or proficiency testing for human SDMA or ADMA; hence, a method comparison was the only option available. Both private and research laboratories offering SDMA and ADMA analysis by ELISA and LC-MS/MS were approached with the hope of organising a sample exchange but unfortunately this wasn’t successful. ELISA kits are known to be expensive, time consuming and error prone; the SDMA ELISA took 5 h to complete with the ADMA ELISA requiring an overnight incubation prior to further analysis. The LC-MS/MS assay is easy to undertake, and a full plate of 96 samples takes less than 1 h to prepare, generating results for SDMA, ADMA and creatinine using only 10 µL of sample within 3.5 h.
Iohexol derived mGFR exhibits a clear relationship with SDMA which mirrors a previous publication, 20 thus highlighting the utility of SDMA as a renal function marker. Studies have suggested SDMA correlates with measured GFR better than creatinine; thus, it may be more useful in detecting early renal dysfunction in male and female patients with low muscle mass or in the paediatric population where subtle changes need to be picked up and acted upon.20,21
SDMA is unaffected by diet, muscle mass, inflammation, oestrogen therapy and diabetes which are all known to have an impact on creatinine and cystatin C concentrations.22–26 In addition to this, obesity, age, gender and polycystic ovary syndrome have a minimal impact on SDMA concentration.27–31 Drug dosing and regimen rely heavily on accurate renal function assessment, further underlining the potential utility of SDMA measurement in patients where creatinine may not be giving an accurate estimation of GFR.
Conclusion
To conclude, we have developed and fully validated a simple and rapid LC-MS/MS method for SDMA, ADMA and creatinine. We believe the addition of an IIV study and assessment of stability both pre- and post-centrifugation prove the analytical suitability of SDMA and ADMA, which to date seem to have been under-utilised in clinical monitoring and diagnosis in humans.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Successful at REC review, IRAS No. 273368.
Guarantor
DJM.
Contributorship
The development and validation of the assay was performed by DJM; sample comparisons were performed by DJM. All authors contributed to the manuscript.