
Abstract
Combined or mixed hyperlipidaemia is characterised by hypercholesterolaemia together with high triglyceride concentrations. It is found in approximately 1 in 100 people in the United Kingdom. Most cases are secondary to an underlying condition such as the metabolic syndrome, diabetes mellitus (especially poorly controlled) or individuals with a high alcohol intake. Mixed hyperlipidaemia is also a feature of some primary hyperlipidaemia conditions such familial combined hyperlipidaemia (FCH) or type III hyperlipidaemia (dysbetalipoproteinaemia). One differential diagnosis for mixed hyperlipidaemia that can easily be overlooked is a patient with an underlying diagnosis of familial hypercholesterolaemia (FH) who also has a hypertriglyceridaemia due to any other cause. Those patients may have very high total and low-density lipoprotein cholesterol concentrations (LDL-C) with a moderately elevated triglyceride concentration. In this article, we report 4 cases of familial hypercholesterolaemia, confirmed by genetic testing, in patients initially presenting with hypertriglyceridaemia in addition to high total cholesterol and LDL-C. This article discusses the diagnostic challenges associated with this presentation and highlights the key role of directly measuring LDL-C to aid diagnosis in these specific situations.
Introduction
Familial hypercholesterolaemia (FH) is a group of inherited genetic defects that lead to the severe elevation of serum cholesterol concentrations. 1 The current clinical practice 2 to categorise the causes of hyperlipidaemia based on which component(s) of the lipid profile is raised has the potential to miss the diagnosis of FH in a patient when their lipid profile also shows a high triglyceride concentration (usually above 4.5 mmol/L) 2 since FH tends to be excluded as a cause of hyperlipidaemia with such a finding. This potentially exposes the patient to risks of delayed diagnosis and failed treatment which, in turn, might lead to premature cardiovascular disease (CVD). An obvious family history of CVD in some of these patients may point towards an inherited hyperlipidaemia; however, in many cases, this is inconclusive, unavailable or may be attributed to the mixed lipid picture.
The finding of high LDL-C in these patients is therefore crucial to alert the clinician to the possibility of an FH diagnosis. However, the result for LDL-C is often unavailable in patients with hypertriglyceridaemia if the clinical laboratory uses the Friedewald 3 or the original Martin-Hopkins formula 4 to estimate the LDL-C concentration. This is because neither equation can reliably calculate LDL-C if the triglyceride concentration exceeds 4.5 mmol/L. By comparison, LDL-C calculated using the more recent Sampson-NIH equation 5 and the updated Martin-Hopkins 6 approach have both been found to be robust up to triglyceride concentrations of 9 mmol/L.
Summary of the 4 cases’ clinical findings.
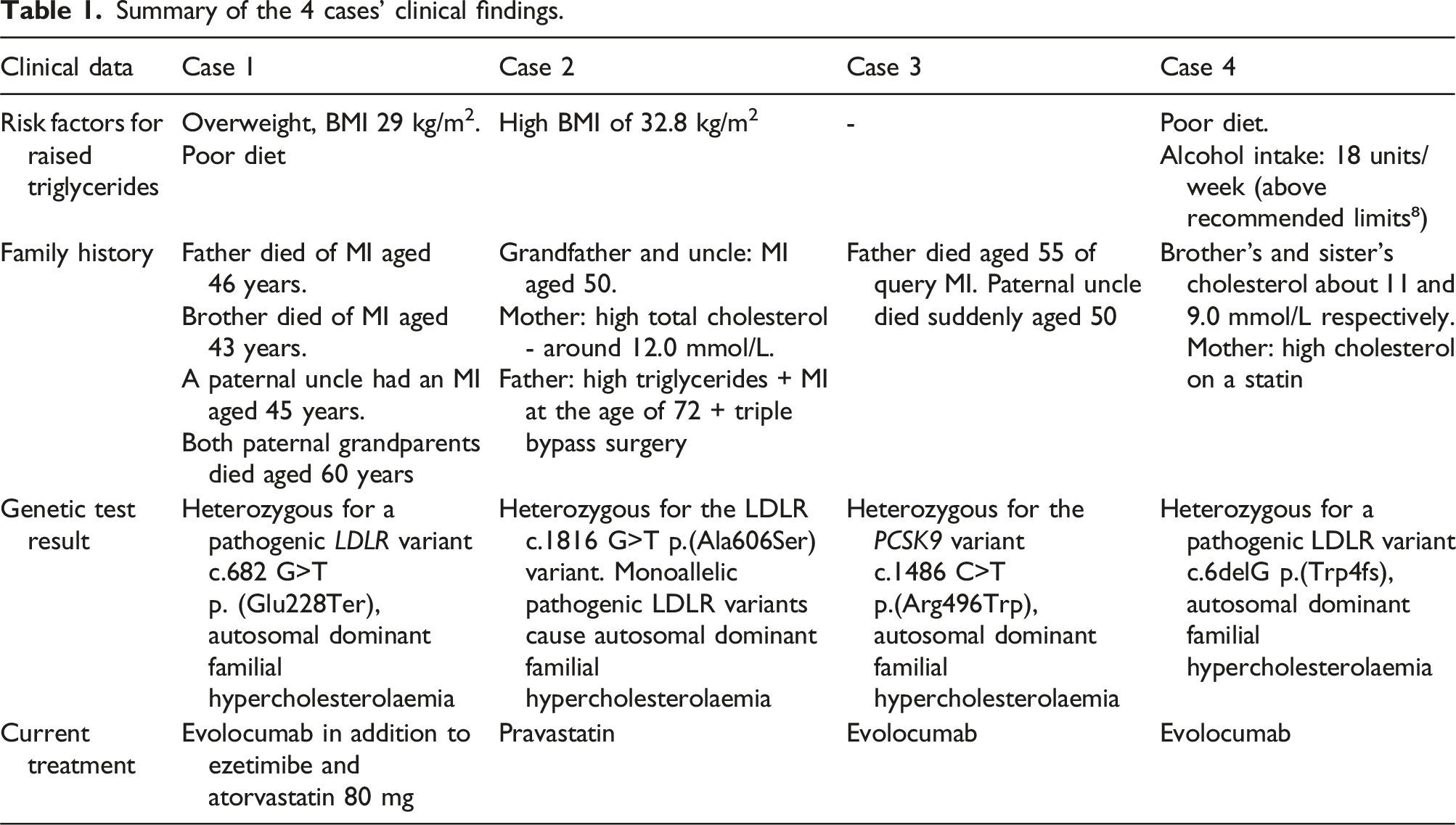
Case 1
Lipid profiles on presentation (results in mmol/L).

Highest and lowest triglyceride concentrations (in mmol/L).

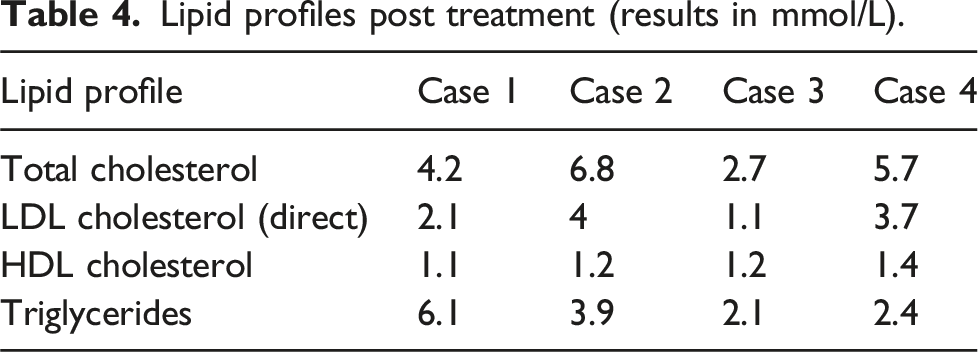
Lipid profiles post treatment (results in mmol/L).
Case 2
A 60-year-old female was diagnosed with mixed hyperlipidaemia and fatty infiltration of the liver. Her lipid profile is shown in Table 2. Her highest and lowest triglyceride concentrations are shown in Table 3.
It proved impossible to obtain a calculated LDL-C concentration on this patient due to the high triglyceride concentration. Her Apo E genotype was not consistent with type III hyperlipidaemia.
Her family history was significant (Table 1) as her grandfather and uncle experienced MIs at the age of 50 years and her mother had been hypercholesterolaemic at around 12.0 mmol/L before treatment with a statin. Her father was also hypertriglyceridaemic and had an MI at the age of 72 years requiring triple coronary artery bypass surgery. She was initially diagnosed with familial combined hyperlipidaemia (FCH) which was reviewed later with the availability of genetic testing for FH. The result reported that this patient was heterozygous for the LDLR c.1816G>T p. (Ala606Ser) variant. Monoallelic pathogenic LDLR variants cause autosomal dominant familial hypercholesterolaemia. This variant is considered to potentially be a mild pathogenic variant or a susceptibility variant which in combination with other factors may lead to hypercholesterolaemia. Therefore, the result was considered to be consistent with a diagnosis of familial hypercholesterolaemia with variable phenotype and reduced penetrance.
She was obese despite her diet being almost vegetarian and including minimal alcohol. However, although active, this was limited as she had multiple sclerosis. She did not have diabetes and was euthyroid. Regarding lipid lowering treatment, she could not tolerate simvastatin or atorvastatin due to muscle aches and joint pains but was able to tolerate pravastatin. Her lipid profile showed some response as shown in Table 4.
Case 3
A 45-year-old female was first seen with a mixed hyperlipidaemia and lipid profile values as shown in Table 2. Her highest and lowest triglyceride concentrations are shown in Table 3. Her father had died at the age of 55 years from what was likely an MI, although no definite diagnosis was made. Her paternal uncle died suddenly around 50 years of age. She had five siblings who were all well. It was initially felt that her family history was inconclusive for FH and in this individual the LDL-C result was unavailable due to a hypertriglyceridaemia. She had a normal BMI and drank no alcohol. There was no obvious cause for her hypertriglyceridaemia. An Apo E genotype test was requested and the result did not support the diagnosis of type III hyperlipidaemia.
She was reviewed again and a decision to perform a genetic test for FH was taken because of her high LDL-C identified following the availability of directly measured LDL-C. Her genetic test for FH was reported as positive with a PCSK9 variant c.1486C>T p. (Arg496Trp).
She was unable to tolerate statins and was subsequently started on evolocumab, to which she responded well as shown in Table 4.
Case 4
A 43-year-old male presented with mixed hyperlipidaemia as shown in Table 2. His highest and lowest triglyceride concentrations are shown in Table 3. His brother and sister’s total cholesterols were markedly raised, being around 11 mmol/L and 9.0 mmol/L, respectively. His mother was also initially hypercholesterolaemic before taking a statin.
In view of the family history and high directly measured LDL-C, he was diagnosed with possible FH and a genetic test requested. The result showed that he was heterozygous for a pathogenic LDLR variant c.6delG p. (Trp4fs).
He admitted to having a poor diet and his average alcohol intake was 18 units a week. He was unable to tolerate statins and was eventually started on evolocumab after which the lipid profile improved as shown in Table 4.
Discussion
Given the prevalence of combined or mixed hyperlipidaemia, the presence of hypertriglyceridaemia in patients with FH may cause diagnostic difficulty as hypertriglyceridaemia is not an uncommon feature of secondary hyperlipidaemias. A review article by Cuchel et al. that discussed the diagnosis and management of FH stated that the condition is often underdiagnosed and undertreated, and that the presence of high triglycerides can complicate the diagnosis, as it may suggest other causes of hyperlipidaemia, such as the metabolic syndrome, diabetes or excess alcohol consumption. 7
Combined or mixed hyperlipidaemia is characterised by hypercholesterolaemia and high triglyceride concentrations and is commonly acquired secondary to conditions such as those already just mentioned. Primary causes of mixed hyperlipidaemia include familial combined hyperlipidaemia (FCH) and type III hyperlipidaemia. Whilst Apo E genotype can be used to exclude type III hyperlipidaemia, the underlying genetic basis of FCH is not known, with it most likely a polygenetic disorder. 10
FH, on the other hand, is a genetic disorder caused by mutations in the genes related to the low-density lipoprotein receptor (LDLR) pathway and is transmitted by autosomal dominant inheritance. Patients with FH have markedly raised LDL-C and usually triglycerides are within the normal range. In contrast, patients with a mixed hyperlipidaemia have an elevated triglyceride concentration 11 which is generally due to an increase in triglyceride-rich very low-density lipoprotein (VLDL) particles and so the total cholesterol concentration is usually only moderately elevated. However, this case report identified four patients presenting with high total cholesterol and triglyceride concentrations found to have a genetic diagnosis of FH. Apo E genotype was performed to exclude type 3 hyperlipidaemia and found to be negative in the first three cases. In case number 4, hypertriglyceridaemia was attributed to a high alcohol intake. So whilst the likely cause for the hypertriglyceridaemia was identified in some of these cases, others had no obvious cause.
The strong family history of CVD and/or the availability of LDL concentration measurement at presentation have helped to point towards the diagnosis of FH earlier in 2 of the 4 cases meaning the diagnosis was not missed.
All these cases emphasise the importance of not attributing every mixed hyperlipidaemia simply to possible secondary causes and associated lifestyle factors. This is important as heterozygous FH is relatively common, with its prevalence estimated as being between 1 in 250 and 1 in 500 of the UK population. 11 Despite these theoretical estimated prevalences, fewer than 1% are diagnosed in most countries. 11
The identification of FH is crucial as this will define patients with high CVD risk who will need treatment for their hyperlipidaemia to reduce the risk of cardiac events. It will also potentially lead to tracing of possible family members also affected. Family history of CVD in some patients is so evident as to trigger applying the Simon Broome criteria and diagnosing FH; however, in many cases, family history is inconclusive or unavailable.
Decisions on how to manage the hyperlipidaemia can also be directly affected by LDL concentration. For example, in cases 2 and 3 neither of the patients would have been eligible for PCSK9 inhibitor treatment according to NICE guidelines if the diagnosis of FH had not been confirmed. 12
These findings also highlight the potential usefulness of direct LDL-C measurement. The initial result for calculated LDL-C was unavailable in three of these patients due to hypertriglyceridaemia. Some laboratories have now introduced a direct LDL-C assay to measure the actual concentration which enables detection of a possible FH diagnosis despite high triglycerides. However, in the United Kingdom, LDL-C is still predominantly estimated by calculation based on the measurement of total cholesterol, HDL cholesterol and triglycerides: in a recent UK NEQAS external quality assessment scheme report, only 61 out of the 524 laboratories participated in the LDL cholesterol scheme were obtained by a directly measured LDL-C assay, with the remainder being obtained using the calculated Friedewald method. 13 Since this equation is unreliable if the patient is non-fasting or has a triglyceride concentration beyond 4.5 mmol/L, direct LDL-C assays may prove useful in this situation in distinguishing increased LDL from VLDL cholesterol when calculated LDL-C is not valid. Whether more recent LDL-C formulae such as Sampson-NIH or updated Martin-Hopkins can serve a similar purpose requires further investigation.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This paper has originated from Manchester University Foundation Trust.
Ethical approval
Informed consent was obtained from all patients and documented in their hospital medical records.
Guarantor
None.
Contributorship
All authors have contributed to and approved the final article.