
Abstract
Background
Ketamine is an NMDAR antagonist with aggregating use across many areas of medicine. P450 enzymes heavily metabolise ketamine, where norketamine is a first pass formed metabolite following initial N-demethylation. Serum ketamine monitoring is becoming increasingly important, requiring a sensitive method with a robust lower limit of quantitation.
Methods
Samples were prepared using protein precipitation or solid phase extraction. Ion suppression was investigated to optimise sample preparation technique, followed by reverse-phase chromatography coupled with tandem mass spectrometry to analyse extractions using a Waters Xevo TQ-S Micro and associated Acquity chromatography systems. Performance characteristics were analysed to validate the assay.
Results
Ketamine and norketamine retention times were 1.28 and 1.23 min, respectively. Ketamine and norketamine precursor ions fragmented into 2 distinguishable product ions (238.14 > 207.18/125.06 and 224.1 > 178.96/124.86). Performance characteristics include an assay recovery of 103.7% (ketamine) and 96.3% (norketamine), lower limit of quantitation 36.2 µg/L (ketamine) and 38.9 µg/L (norketamine), and intra-assay imprecision ≤ 7.04% on average.
Conclusions
A robust and reproducible assay with limited sample preparation has been designed and validated. The linearity of the assay covers all ranges of interest reported in the literature. Ion suppression was clearly reduced via use of solid phase extraction. The method will form the basis of ketamine monitoring and providing valuable patient information on tolerance and metabolism.
Keywords
Background
Ketamine is an anaesthetic drug with an increasing clinical use. 1 It is clinically and pharmacologically unique in simultaneously producing hypnotic, amnesic and analgesic outcomes.1,2 There is an accumulating use across medicine of ketamine to induce dissociative anaesthesia and relieve chronic and acute pain, as well as emerging evidence in facilitating abstinence in substance addiction.2,3 Ketamine has furthermore demonstrated depressive symptom and suicidal thought reduction properties and supporting status epilepticus.4,5 Consequently, establishing a laboratory method for future definition of therapeutic concentrations and targeted plasma Cmax levels is essential in optimising patient care. The phencyclidine derived (R,S)-ketamine exists as a racemic compound of molecular weight 237.72. As two optical enantiomers, R and S, ketamine is used either in racemate or as the S-ketamine stereoisomer, known commercially as Esketamine.1,6,7
Different types of administration endorse a clinical laboratory method with good analytical performance throughout a wide concentration range. Ketamine can be administered parenterally or orally, depending on symptoms targeted, patient age and tolerability; examples include IM in paediatrics, IV in chronic pain relief and oral formulations in adjuvant pain therapy.8,9 IV administration is preferable to achieve total bioavailability. 9 Oral administration is hindered by hepatic metabolism, limiting the bioavailable ketamine, although sublingual wafers have been explored to evade the liver and increase bioavailability. 10 Administration and dose variants require support from the laboratory via robust monitoring, where serum concentrations are hypothesised to fluctuate between alternative administration techniques. Consequently, a method to monitor ketamine must have a broad linearity.
Ketamine is lipid and water soluble, reaching target tissues rapidly where the volume of distribution is 3 L/kg at steady-state. 11 The extent of protein binding has been disputed in the literature, ranging from 10–50%. 12 Furthermore, plasma Cmax levels for symptom relief can coincide with side effects. For example, analgesia has been displayed at plasma concentrations between 70 and 160 µg/L, while psychosis has been reported at 120 μg/L.1,13,14 A harmonised ketamine target range is a chief interest in optimising patient care. Discrepancies in unbound ketamine and the high volume of distribution could indicate a narrow therapeutic window, supporting the need for laboratory monitoring of serum/plasma ketamine concentrations. Potentially low peripheral concentrations following the high volume of distribution, unknown protein binding and lipid solubility necessitate a method with good lower limit of quantitation. Consequently, the ideal approach to quantitating serum ketamine is an ultra-high-performance reverse-phase liquid chromatography tandem mass spectrometry (UPLC-MS/MS) method paired with a sample preparation to remove interferents and minimise ion suppression while maintaining signal and analyte recovery.
Principally, ketamine antagonises N-methyl-D-aspartate receptors (NMDAR). This dynamic is attainable as ketamine passes the blood–brain barrier to the central nervous system freely. Ketamine reduces glutamate activity in the synaptic cleft, desensitising response to stimuli and pain. As dose increases during titration of IV, ketamine may bind to a second site in the NMDAR and decrease channel opening, further depolarising the synapse. Hence, increasing dose improves ketamine’s efficacy, especially useful in achieving long-term chronic pain relief. Monitoring ketamine at these higher doses is significant to the clinician in avoiding toxicity and maintaining patient experience. Neurotoxicity is a distinct possibility at high doses, as apoptotic lesion promotion may occur. Current monitoring at this level relies on one-to-one patient observation for side effects such as nausea, nystagmus and psychosis. This could be avoided by using data generated from robust laboratory monitoring.1–3,13–15
Ketamine metabolism is extensive. Nitrogen demethylation to norketamine is catalysed hepatically by P450 enzymes; chiefly CYP3A4 and to a lesser extent by CYP2B6 and CYP2C9. 16 The catalysis is stereoselective where S-ketamine is modified faster by CYP3A4 than R-ketamine. 1 Norketamine exhibits 80% of the potency of the parent compound ketamine and is therefore a prime active metabolite for monitoring purposes. P450 inter-subject variability explains pharmacokinetic disparity in ketamine metabolism and is partially to blame for tolerability differences.17,18 Isolated P450 enzymes incubated with R and S ketamine in vitro have formed varying ‘fingerprints’ of ketamine metabolites. These include, but are not limited to, hydroxylation of norketamine to hydroxynorketamine by CYP2A6 and dehydrogenation of norketamine by CYP2B6 to dehydroxynorketamine. 17 Therefore, measurement of ketamine in therapeutic monitoring would not benefit from measurement of any metabolites past norketamine, as further metabolites have significant variability. Likewise, stereoselective measurements would not benefit the clinical laboratory setting where racemates are still commonly used in drug formulations.
We present a UPLC-MS/MS method development for total racemic ketamine and norketamine measurement. The method has proven suitable for measuring serum concentrations of total S-ketamine, S-norketamine, R/S-ketamine and R/S-norketamine in patients receiving IV and oral formulations for chronic pain. The method has been validated to published criteria. 19
Methods
Materials and methods
Equipment
Equipment used included a Waters™ Positive Pressure-96 Processor and Waters™ Xevo TQ-s Micro with Acquity Ultra-High-performance chromatography (UPLC) systems. A Waters Acquity UPLC® BEH C18 column was used for chromatographic separations.
Chemicals and reagents, standards and quality control materials
Certified reference materials of ketamine and norketamine, and stable-labelled analogues of both compounds (deuterium = 4) manufactured by Cerilliant, were purchased from Merck. HPLC-grade acetonitrile, water and formic acid (98% w/w) were purchased from VWR International. ACQ Science DCT-SE QC material was purchased via LGC. Anhydrous Zinc sulphate heptahydrate, phosphate buffered saline (PBS) and bovine serum albumin (BSA) were obtained from Sigma-Aldrich. Ostro™ Protein Precipitation & Phospholipid Removal Plates and 2 mL 96 well plates were purchased from Waters™. Mobile phase A utilised HPLC-grade water with 0.2% formic acid. Mobile phase B utilised HPLC-grade acetonitrile with 0.2% formic acid. Acetonitrile was chosen for a higher elution strength and anticipated short retention times. Formic acid was added for ionisation enhancement to gain the signal needed at the hypothesised low peripheral concentrations of ketamine. Standards were prepared by using a 1 mg/mL solution of ketamine and a 1 mg/mL solution of norketamine. Each solution was diluted to an intermediate = 20,000 µg/L. All dilutions were prepared in PBS/0.1% BSA and both ketamine and norketamine had the same concentrations. 6 standards were prepared by diluting the intermediate standard to 2000 µg/L (standard 5). Standard 5 was serially diluted to 1000 µg/L, 500 µg/L, 250 µg/L and 175 µg/L. The sixth standard was purely PBS/0.1% BSA serving as a ‘zero’. Ketamine and norketamine were spiked into analyte free serum at approximately 300 µg/L and 1200 µg/L to serve as quality control (QC) material. Third Party QC in a serum matrix was available from ACQ Science (DCT-SE). This was pre-assayed at 1010 µg/L and used for accuracy purposes alongside a LGC drug confirmatory whole blood scheme. Internal standards followed two preparations utilising ketamine D4 and norketamine D4 both at 100 µg/ml. For the protein precipitation, an intermediate standard was made at 1000 µg/L in acetonitrile. 1 mL 0.9 M ZNSO4, 7 mL acetonitrile and 2 mL of intermediate standard were mixed for a combined protein precipitation reagent and internal standard. For the solid phase extraction, the same intermediate was diluted 1 in 10 with PBS/0.1% BSA. Both internal standards had working concentrations of both deuterated standards at 100 µg/L.
Sample preparation
Two sample preparation procedures were investigated: solid phase extraction (SPE) and protein precipitation (PP). Ion suppression was subsequently investigated, to determine the better method. Each plate was thermo-sealed prior to LC-MS/MS injection.
Protein precipitation
20 µL of calibrant, QC or sample were combined with 80 µL of PP/internal standard, mixed for 5 min at 700 RPM and centrifuged at 3700 RPM for 5 min. 80 µL of HPLC-grade water was added, mixed for 5 min at 700 RPM and centrifuged at 3700 RPM for 5 min.
Solid phase extraction
100 µL of calibrant, QC or sample were combined with 25 µL of internal standard and 400 µL of elution reagent (acetonitrile with 1% formic acid) in an Ostro Protein Precipitation & Phospholipid Removal Plate and mixed for 5 min at 700 RPM. 5–6 min of positive pressure at 6 psi using Waters™ Positive Pressure-96 Processor applying regulated source pressured nitrogen (100 psi) was used to collect the eluent. The eluent was further diluted adding 1 mL of HPLC-grade water. The collection plate was centrifuged at 3700 RPM for 5 min.
Ultra-performance liquid chromatography
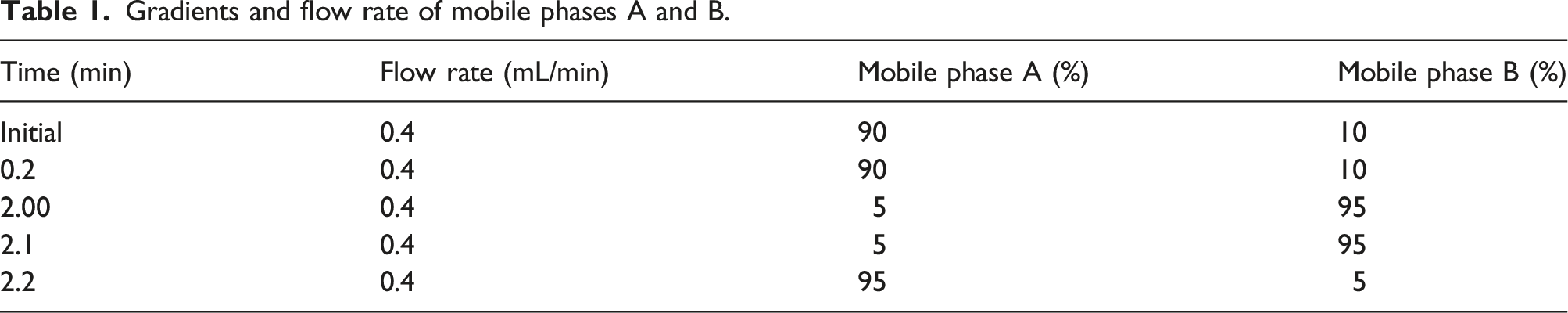
Gradients and flow rate of mobile phases A and B.
Tandem mass spectrometry
Ionisation and subsequent fragmentation were achieved using electrospray ionisation in positive mode. Capillary voltage was maintained at 0.7 kV. MRM was employed to monitor several ions (Figure 1). Chromatograms for ketamine/norketamine ions and deuterated isotopes. Details mass transitions and energies, retention times and ion count for highest calibration standard (ketamine and norketamine = 2000 µg/L). Y-axis = peak intensity (%). X axis = time (minutes).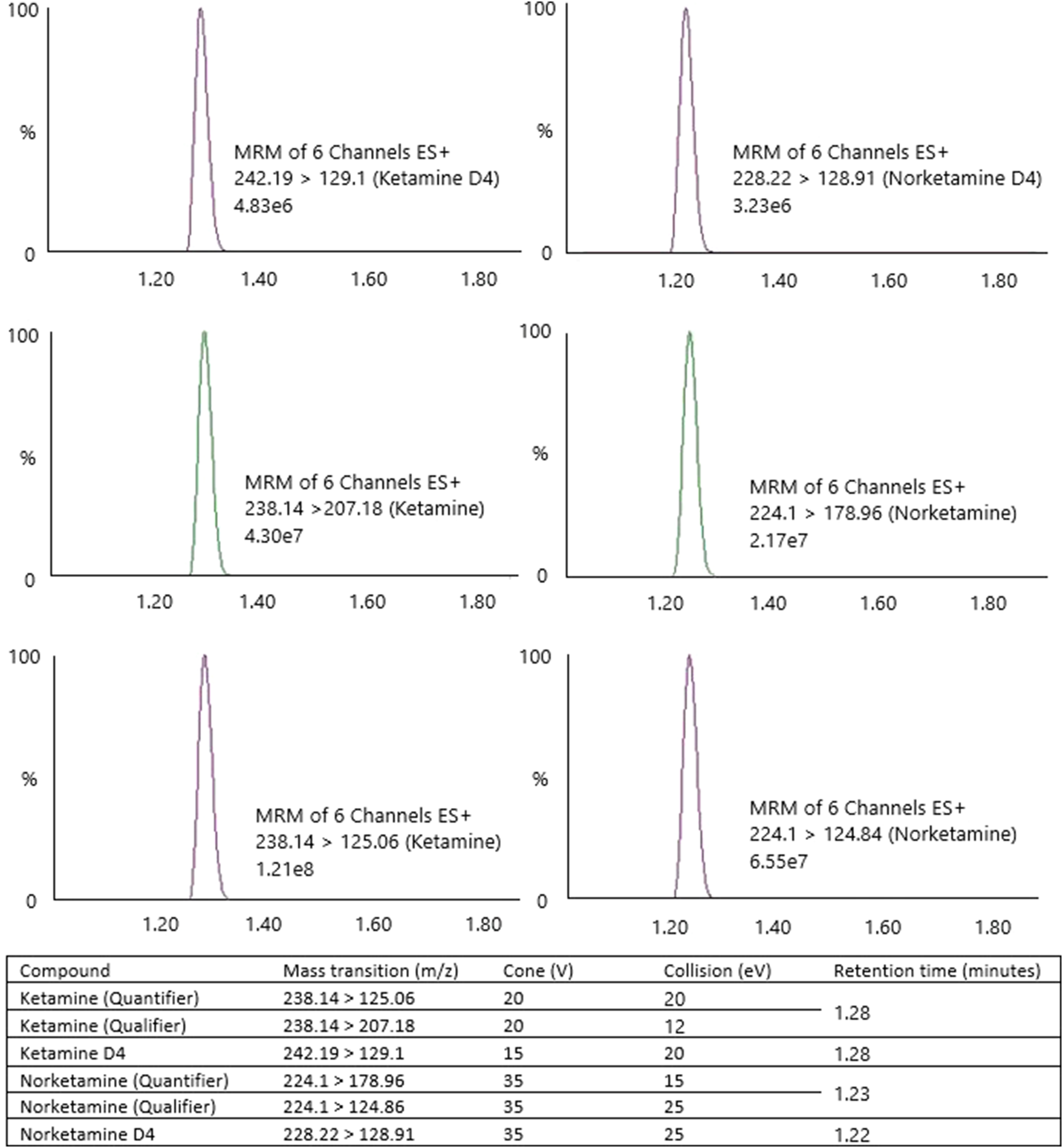
Performance characteristics
Investigations into ion suppression, accuracy and imprecision, recovery, linearity, specificity, and sensitivity were employed following published principles. 19 Samples were donated by the Liverpool Neuroscience Biobank at the Walton Centre from patients known to be receiving ketamine titrations for pain relief.
Ion suppression
Ketamine-free serum samples were extracted by both PP and SPE. No internal standard was added to these samples. Internal standard peaks were super-imposed over the extractions. Ion enhancement or suppression was defined as an intensity change of ≥20%.
Accuracy and imprecision
Samples of patients receiving ketamine treatment were obtained from the Liverpool Neuroscience Biobank at the Walton Centre and measured via the designed UPLC-MS/MS method, correlated with results from a referral laboratory using HPLC-Diode Array Detection (HPLC-DAD). There were no clinical method comparisons available for norketamine. Replicates of QC material 1, 2 and the ACQ Science DCT-SE were measured within and between batch to determine both intra-precision and inter-precision (ketamine only). Targets of coefficient of variation (CV) < 10% were set. No current external quality assurance scheme exists to measure serum ketamine and norketamine. A whole blood toxicology scheme (LGC QUARTZ) and ACQ Science DCT-SE (pre-assayed) were used to assess accuracy for ketamine only. Each third party assayed material was measured in duplicate.
Blinded dilutions of pre-assayed material were also used to assess a theoretical diluted concentration against a measured value. Patient samples were assayed in 2 separate batches, assessing inter-precision for ketamine and norketamine in authentic ketamine-infused patient sera.
Recovery
Known concentrations of ketamine and norketamine were spiked into ketamine-containing serum samples. The theoretical increase in concentration was calculated. The percentage difference between measured and calculated recovery was determined. Recovery targets were set between 90% and 110%.
Linearity
Calibration standards were prepared across a range of 0–2000 µg/L for both ketamine and norketamine. A curve was drawn by producing a response value (response = analyte area/internal standard area). The reproducibility of this standard curve was monitored between batches. Curves were considered linear if the correlation coefficient ≥0.99, calculated using the least squares regression model.
Specificity
A range of interferents was investigated. Available LGC EQA material containing 35 common analgesics, antibiotics, sedatives, antidepressants and anti-epileptics (supplementary item 1) was used as a diluent for a spiked ketamine/norketamine sample. The same sample diluted in PBS/0.1% BSA was analysed adjacent to ascertain any differences between signals and concentrations. Each sample was analysed in triplicate. Common serum matrix interferences were investigated. Standard 5 was diluted in various concentrations of pure bilirubin, intralipid and human haemoglobin and measured to determine whether there were any significant differences (determined as ±10%).
Sensitivity
Lower limit of detection (LLoD) used 3 solutions of ketamine and norketamine approximated at 3.90, 1.95 and 0.97 µg/L. LLoD was defined as the lowest peak producing a signal to noise (S/N) ratio of 3 with a minimum threshold of 3x standard deviation of a blank signal. 19 10 replicates for LLoD were assayed with a target CV <20%. Lower limit of quantitation (LLoQ) was defined as the value with a S/N ratio of 10 producing 10 replicates with a CV <20%. A minimum threshold of 10x the standard deviation of the blank was set. For both LLoD and LLoQ, the final value was calculated by a mean result of 10 replicates +2 standard deviations. Limit of blank (LoB) was analysed using LGC EQA material containing analgesics, antibiotics, sedatives, antidepressants and anti-epileptics but no ketamine or norketamine.
Results
Liquid chromatography tandem mass spectrometry
Both extraction methods gave visibly clear supernatants acceptable for injection into the Acquity UPLC system. Column retention times: ketamine, ketamineD4 = 1.28 min (±0.01) and norketamine, norketamineD4 = 1.22 min (±0.01) (Figure 1). Resolution was not explored further due to MRM use. MRM produced similar intensity peaks for all fragmentations. The detector produced good responses for ketamine and norketamine with peak areas for both compounds between ≈1,000,000–1,300,000. The internal standard produced an average ion count (n = 25) of 88,200 (ketamineD4) and 39,835 (norketamineD4).
Ion Suppression
PP extraction studies indicated high ion fluctuation between 1 and 1.55 min. An increase of 75% signal intensity between 1 and 1.21 min (Figure 2(b)) significantly impacted both ketamine and norketamine, as both compounds eluted near this retention time window. SPE reduced signal fluctuations to <5%, indicating a more stable ion environment for the solvent gradient’s entirety (Figure 2(a)). Due to the ion fluctuations, it was deemed that SPE was a more appropriate sample preparation method. Consequently, SPE accounted for all sample preparation for the performance characteristics. Chromatograms for blank serum, Ketamine and Norketamine. (a) Ion suppression of PP extraction, maximum ion counts = 7.58
4
. (b) Ion suppression of SPE extraction, maximum ion counts = 1.23
4
. (c) Ketamine/norketamine chromatogram. Ketamine RT = 1.28 min, peak ion count = 1.21
8
. Norketamine RT 1.22 min, peak ion count = 6.55
7
. A and B y-axis linked. C y-axis independently scaled.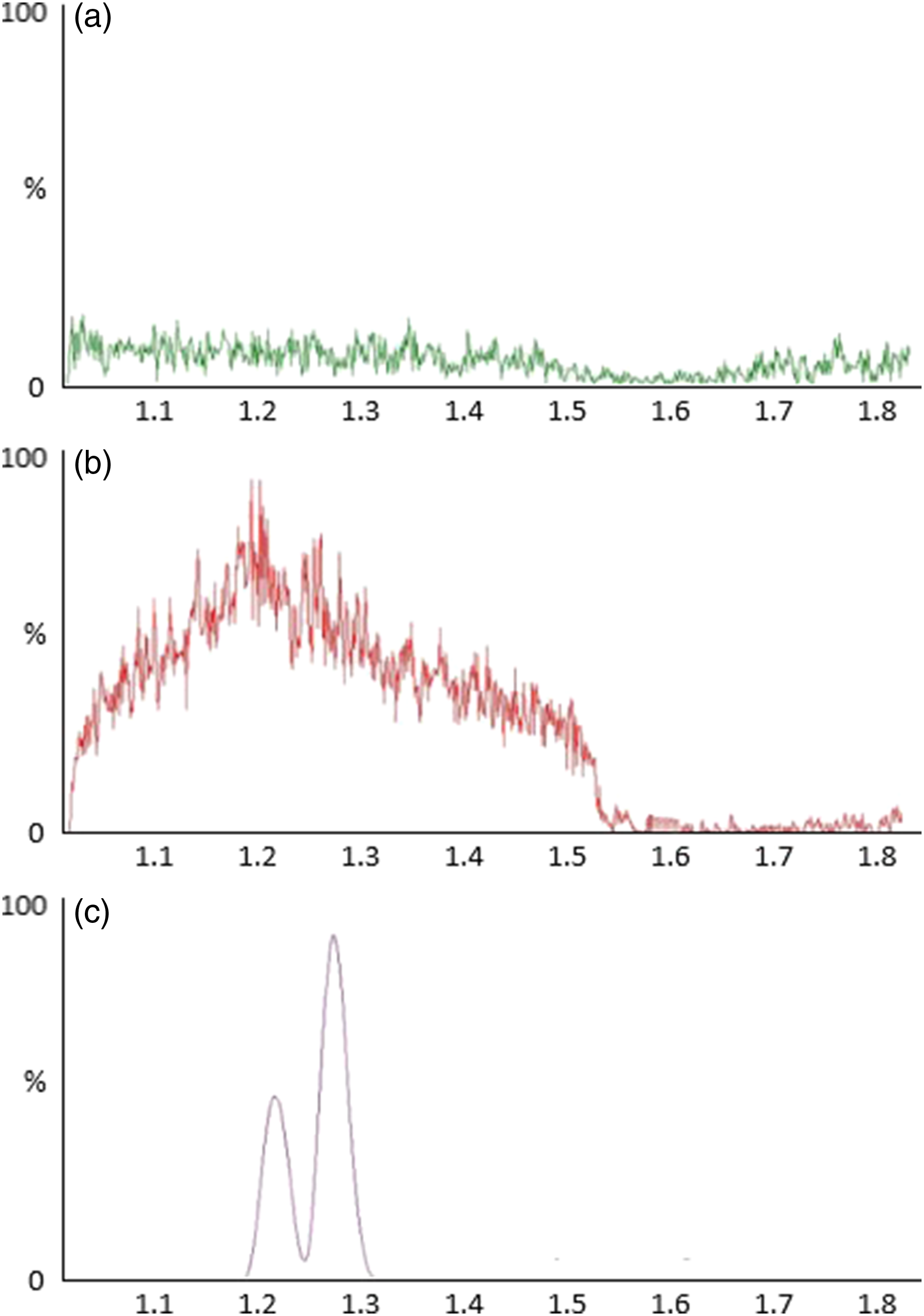
Accuracy and imprecision
Intra-precision was successful where each analyte surpassed target CV (<10%) in 10 replicates: QC1. Ketamine (mean = 270.6 μg/L) 2.5%, norketamine (mean = 345.6 μg/L) 5.5%; QC2. Ketamine (mean = 1182.3 μg/L) 1.5%, norketamine (mean = 1285.6 μg/L) 7.0%. Inter-precision at both QC values averaged at 5.1% (ketamine) and 6.7% (norketamine) (n = 10). ACQ Science DCT-SE gave inter-precision at (3.6%) for ketamine (n = 10) averaging at 1019.9 μg/L (target = 1000.0 μg/L). The assay was deemed precise across the concentration range within and between assays. Samples between batches analysed in duplicate gave an average percentage difference of 6.1% for ketamine and 7.0% for norketamine (n = 15). Method comparison data produced varying results, limited to available sample size. The comparative HPLC-DAD method did not quantify any value <300 µg/L, reducing the number of samples that could be compared between methods (n = 8). Bland–Altman analysis produced a partition bias of +9.08% (range = −27.7% – +47.1%) where a correlation coefficient forced through the intercept = 0.95. Accuracy studies against known third party materials were successful. Partition bias = 20.2 μg/L or 4.6% (95% confidence range 37.9–78.4 μg/L or 9.3–18.4%). No comparative data was available for norketamine. Moreover, norketamine was measurable in all samples (ketamine:norketamine ratio ranged from 0.03 to 2.52. Measurable ranges: Ketamine = 10.8–1287.2 µg/L and norketamine = 70.4–1060.6 µg/L) (n = 23). Further confidence in the presented method can be drawn from Figure 3. Confidence can be drawn from this successful agreement with third party material and the additional blinded dilutions. Bland–Altman plots between third party assayed material and the evaluated method. n = 12. R2 = 0.99. A = absolute differences. Partition bias = 20.2 μg/L. −1.96SD – +1.96SD = −37.9–78.4 μg/L. B = % differences. Partition bias = 4.6%. −1.96SD – +1.96SD = −9.3–18.4%.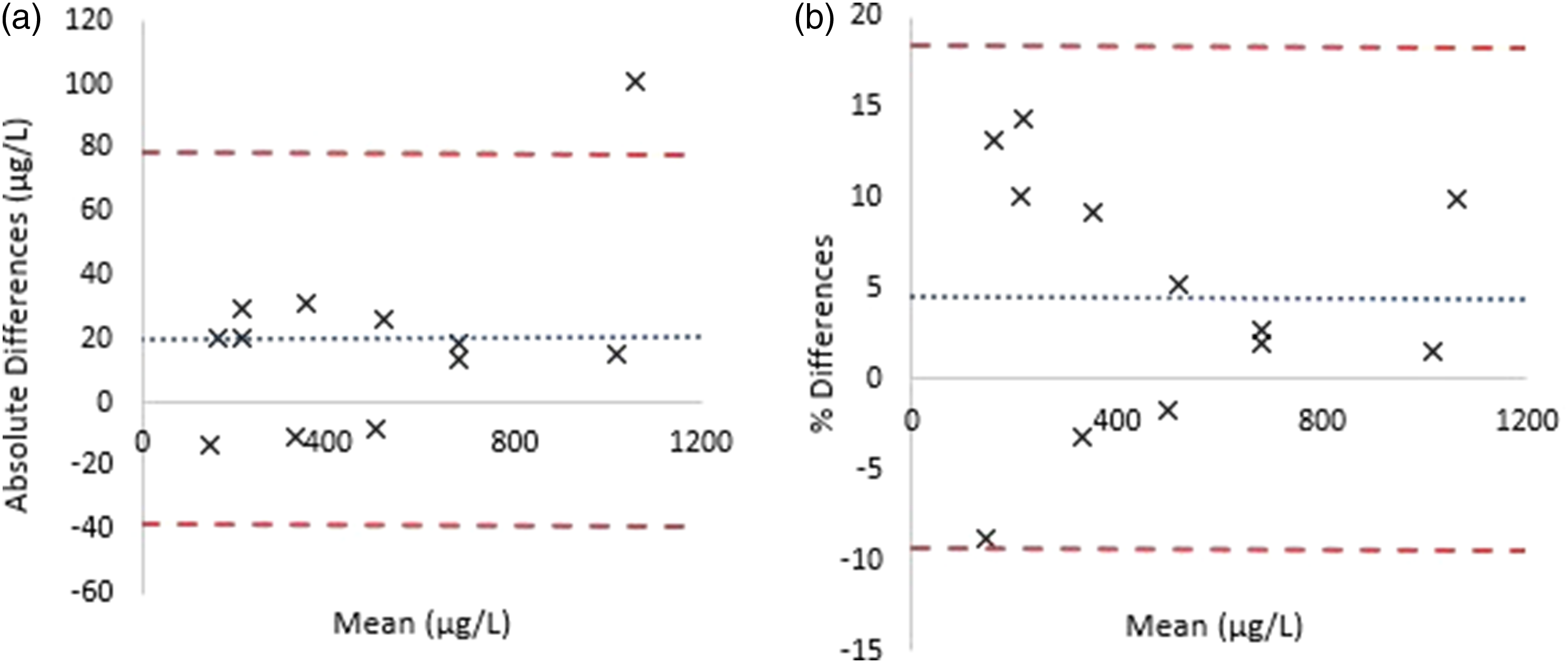
Recovery
Recovery proved an invaluable investigation where method comparisons were limited. Both compounds met target recovery where ketamine recovery = 101.4% (proportional error = −1.4%) and norketamine recovery = 96.3% (proportional error = 3.7%).
Linearity
Standard curves were produced against calibrant values and analyte response (response = peak area * (internal standard area/calibrant concentration)). Regression for both ketamine and norketamine over the ranges 0–2000 µg/L produced good correlations with R2 = 0.9993 (ketamine) and 0.9984 (norketamine).
Specificity
Spiked values into PBS 0.1% albumin and into the complex drug mixture EQA material did not give significant differences (Ketamine: p = .21, Norketamine: p = .49, supplementary item 1). Ketamine was observed not to alter in matrix additives up to haemoglobin 8 mg/dL, bilirubin 6 mg/dL and triglycerides 400 mg/dL with mean biases of 5.2%, 4.2 and −5.9%, respectively. Norketamine did not alter in matrix additives up to haemoglobin 4 mg/dL, bilirubin 6 mg/dL and triglycerides 400 mg/dL with mean biases of 7.6%, −8.4% and 2.4%, respectively.
Sensitivity
Lower limits of performance.
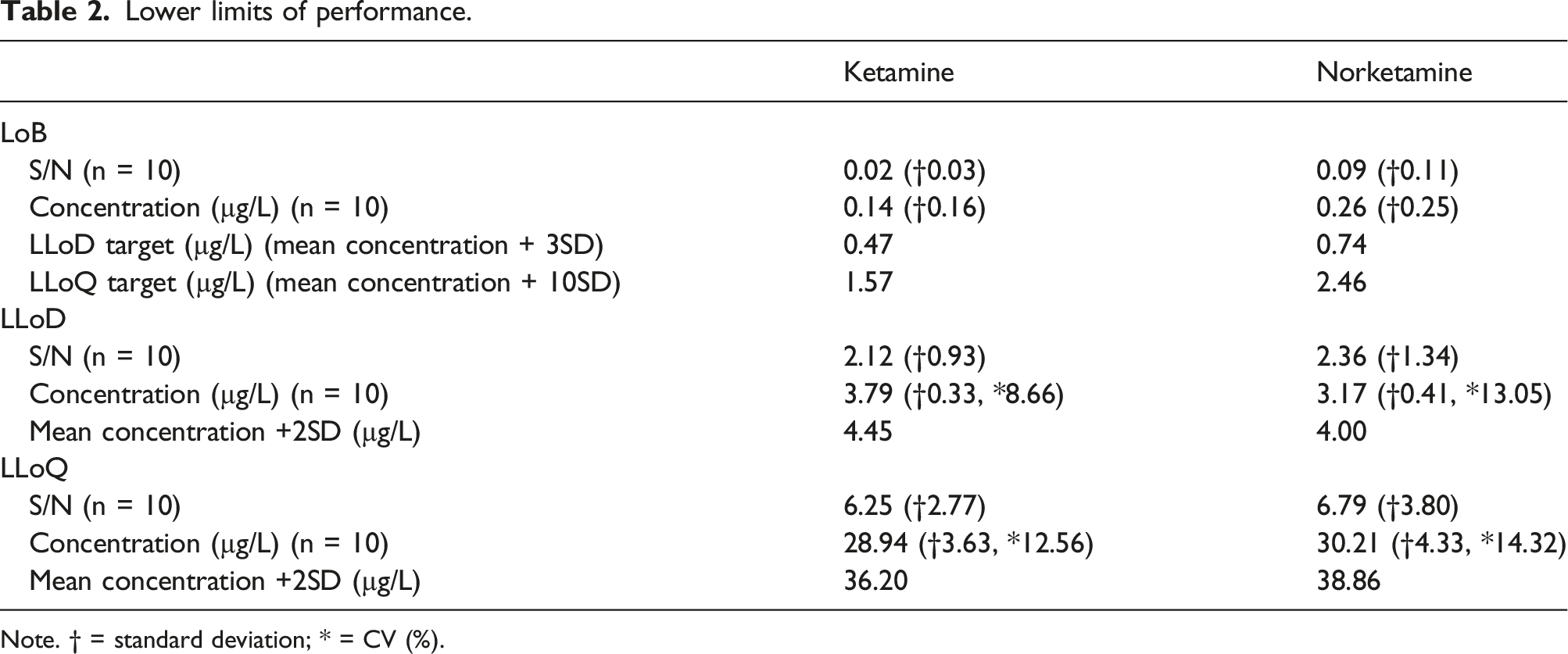
Note. † = standard deviation; * = CV (%).
Discussion
The method developed in this study measured ketamine and norketamine in all patient sera, highlighting the sensitivity needed to support ketamine therapy. There is an increasing clinical use of ketamine at lower dosages to target depression and inflammation (Plasma Cmax≈185 µg/L) 1 and need for analysing trough levels following titrations to relieve chronic pain. 2 Current serum or plasma assays in the UK utilise HPLC-DAD where reporting limits are <300 µg/L. 20 Improvements by this study’s method made to current clinical monitoring are now demonstrated, where the present study ketamine LLoQ was calculated at 36.2 µg/L. Norketamine is not currently reported in therapeutic monitoring, although a LLoQ of 38.9 µg/L was achieved in this study. The LLoQ achieved means the assay linear range for ketamine of 36.2–2000.0 µg/L covers most reported plasma Cmax.1,2,16 Moreover, patient samples were accessed from a wide range of ketamine applications including mid and end titrations and oral top ups, wherein ketamine and norketamine were quantified in all. LLoD results were lower than non-LC-MS/MS methods for ketamine. HPLC-UV is the most common method applied to blood fractions. UV as a detection method produces LLoD ranges 5–12.5 µg/L in methods using SPE as sample preparation. 6 Improvements are most likely due to the sensitivity of tandem mass spectrometry. On the contrary, the reported LLoQ is generally higher than many methods in the literature using a range of chromatography. 6 A similar study using targeted mass spectrometry produced a LLoQ at 0.8 µg/L (S/N target = 10). Sample preparation differences included ultrafiltration and vacuum drying. A silica-based column gradient elution achieved via ultra-high performance micro-flow liquid chromatography (micro-UPLC) gave much longer retention times than the present study, at 4.6 min and 4.8 min for norketamine and ketamine, respectively. 21 A similar study utilising micro-UPLC achieved a LLoQ at 4 µg/L (retention times 7–7.7 minutes) using a micro C18 column. 22 The disparity in LLoQ is most likely due to limited noise contribution in S/N ratios. Operating at high pressure, micro-UPLC using silica-based columns functioning at acidic pH will retain basic compounds, such as ketamine longer. ACQUITY UPLC M-Class HSS T3 columns for micro-UPLC improve desolvation, extending retention times and ultimately improving analyte sensitivity. 23 The rapid elution using a generic BEH C18 column in this study may contribute to the noise noted and increasing the background signal at low concentrations. Hence, the current method could not replicate limits of analysis achievable by micro-UPLC. Using micro-UPLC to improve the current method would not benefit the clinical setting. Furthermore, the comparative micro-UPLC studies linear ranges ended at 201.4 µg/L (ketamine), 200.6 µg/L (norketamine) 21 and 500 µg/L (ketamine and norketamine). 22 This does not cover the appropriate plasma Cmax targets. Column overload at concentrations over the top standard would most likely occur using micro-UPLC, hypothesised by slow column transfer causing misshaped peaks. 24 Run times for the current method are also quicker compared with the published methods.21,22 The current UPLC-MS/MS method is more suitable to the clinical setting.
A limitation of the current study is the lack of chiral separation. Although it is assumed that total ketamine in each sample measured is attributed to the prescribed dose, the current method cannot totally account for the contribution of either R or S-ketamine. Ketamine’s dissociative anaesthetic property is popular amongst drug users. Through 2017–18, 0.8% of UK adults aged 16–59 had used ketamine that year; a 141,000 increase in individuals on the year prior. 25 Ketamine is commonly abused, and most street-drug formulations are racemic, made illegally and cheaply. 26 A method verified in mice has utilised chiral separation of ketamine and norketamine in plasma, cerebrospinal fluid (CSF) and brain tissue detected by MS/MS. 27 Positives proven by this chiral method include low sample volumes, simple deproteinization preparation and good performance characteristics. A CHIRALPAK AS-3R column with run time of 5 min showed LLoQ at 1 µg/L and 2 µg/L of ketamine in plasma and CSF, respectively. Each enantiomer was able to be quantified in this sensitive manner individually where each compound was resolved between 3.2 and 3.6 min. 27 However, the narrative remains the same for a clinical monitoring method: Does stereoselection influence the quality of ketamine monitoring? Stereoselective analysis in the clinical laboratory is not of practical use. Differences between samples collected before and after ketamine infusion would account for the total enantiomer or racemate. The chiral method does bring the question of CSF ketamine measurements. The CNS is highly perfused, and the lipid solubility of ketamine and free transfer to CSF poses the prospect of targeting individual ketamine titrations to CSF measurements and the possibility of alternate ketamine metabolomics in the CNS. Succinctly, CSF measurements of ketamine may be explored through further work and additional validation of this method.
Protein precipitation performed poorly, enhancing ion response up to 75% at the retention time windows for both compounds of interest. As per performance characteristic targets, this enhancement in ion signal prompted the choice of sample preparation as solid phase extraction. Solid phase extraction performed well recovering both ketamine and norketamine at 101.4% and 96.3%, respectively. Reported recovery bettered previously reported overall recovery for comparable extraction investigation of ketamine in analysis in pig plasma (overall recovery ranged 80–89%). 28
Specificity has been displayed in this method. Sharp peaks were produced with no enhancement or suppression in signal for common serum matrix interferents or the 35-drug matrix ketamine/norketamine spiked samples. The literature has been explored, and although crude, no other study has investigated as many possible drug assay interferents with a serum ketamine or norketamine method. Known drug interactions with ketamine investigated include alprazolam, clonazepam, gabapentin, lamotrigine, levetiracetam and midazolam (CNS depressant effects), phenobarbital, phenytoin, primidone, and carbamazepine (decreased exposure and adjusted dose required) and theophylline (severe risk and increase in seizures). 29 Polypharmacy patients on any of the stated drugs and ketamine can still achieve reliable levels following measurement by the current method. In addition, intra- and inter-assay performance is satisfactory across the linear range. Relative standard deviations expressed as %CV were comparable across most methods. Intra-assay average CV for both QC values was 2.0% (ketamine) and 6.3% (norketamine) compared to reported 2.6% and 10.0% (ketamine) and 3.0% and 8.1% (norketamine).27,29 Inter-assay average CV for both QC values was 5.7% (ketamine) and 6.1% (norketamine) compared to reported 8.4% and 4.3% (ketamine) and 9.0% and 3.2% (norketamine).27,29
General correlations ensued with method comparison data where linear regression = 0.95. While partition bias averaged at +9.08%, the data are truly reflected by the limits of agreement range at −27.7% – +47.1%. There was limited agreement in the methods. HPLC-DAD and UPLC-MS/MS should be used separately. An in-depth comparison between HPLC-DAD and UPLC-MS/MS concluded recoveries between the separate detection methods were statistically different when measuring tetracycline in the same extraction matrix (Fisher-Snedecor = 0.05). 30 Further differences have been illustrated in a similar method comparison where lamotrigine is the target compound, which like ketamine has a 2 aromatic ring structure. Differences ranged from −32.0% to +48.6% and regression = 0.97, like the current study. 31 Confidence in our method is demonstrated as all performance characteristics were met.
Conclusion
The analysis of ketamine and norketamine in blood is of clinical value in a variety of settings. Chiefly, the present study results met the initial aims. A robust, rapid, reproducible, sensitive and specific method was developed and validated. Other highlights include simple sample preparation, short run times and relatively inexpensive running costs. Although the initial capital required for LC-MS/MS systems is high, reagent and consumable prices are relatively low in comparison to commercially available chemistry assay kits and platforms. LC-MS/MS platforms will remain resilient if properly maintained and used appropriately. Performance for limits of analysis was higher than initial estimations. However, it is believed the assay measuring range will cover all clinically appropriate concentrations the laboratory may encounter. Further work may be centred around pharmacokinetics, stability, protein binding and improving limits of analysis. Currently, chiral separation is not necessary in our clinical setting, as our Trust is moving to the use of Esketamine.
Supplemental Material
Supplemental Material - A rapid, sensitive method for clinical monitoring of ketamine and norketamine by ultra-high-performance reverse-phase liquid chromatography tandem mass spectrometry
Supplemental Material for A rapid, sensitive method for clinical monitoring of ketamine and norketamine by ultra-high-performance reverse-phase liquid chromatography tandem mass spectrometry by Nicholas Armfield, Bernhard Frank, and Carrie Chadwick in Annals of Clinical Biochemistry.
Footnotes
Acknowledgements
Thank you to Dr Sally Hanton manuscript preparation guidance. Thank you to Dr Ioannis Kanakis for supporting the project. Thank you to all staff at the Walton Centre Neuroscience Laboratories for support.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by The Walton Centre NHS Foundation Trusts.
Ethical approval
The ethics committee of The Liverpool Neuroscience Biobank at The Walton Centre approved this study (Ethics Reference LNBW 22_03).
Guarantor
Nicholas Armfield.
Contributorship
Project formulation by Dr Bernhard Frank and Carrie Chadwick (The Walton Centre NHS Foundation Trust). Principal investigations, method design, lab. work, statistics, figures, and text for the manuscript prepared by Nicholas Armfield.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.