
Abstract
Alkaptonuria is an iconic rare inherited inborn error of metabolism affecting the tyrosine metabolic pathway, resulting in the accumulation of homogentisic acid in the circulation, and significant excretion in urine. Dating as far back as 1500 BC in the Egyptian mummy Harwa, homogentisic acid was shown to be central to the pathophysiology of alkaptonuria through its deposition in collagenous tissues in a process termed ochronosis. Clinical manifestations occurring as a consequence of this are typically observed from the third decade of life, are lifelong and significantly affect the quality of life. In large supportive and palliative treatment measures are available to patients, including analgesia, physiotherapy and joint replacement. Studying the natural history of alkaptonuria, in a murine model and human subjects, has provided key insights into the biochemical and molecular mechanisms underlying the pathophysiology associated with the disease, and has enabled a better understanding of the common disease osteoarthritis. In the last decade, a major focus has been on an unlicensed disease-modifying therapy called nitisinone. This has been shown to be highly efficacious in reducing homogentisic acid, and it is hoped this will halt ochronosis, thus limiting the clinical complications associated with the disease. A well-documented metabolic consequence of nitisinone therapy is hypertyrosinaemia, the clinical implications of which are uncertain. Recent metabolomic studies have helped understand the wider metabolic consequences of nitisinone therapy.
Case vignette
It was back in 2000 that Nick Sireau first heard about the ultra-rare disease alkaptonuria (AKU). His first son had just been born and he noticed that his nappies were staining red-black. An emergency doctor said it was dye from the red cabbage his wife had eaten that was somehow getting into the breast milk and into the baby.
They could not believe this, so went to see their GP who eventually diagnosed it as AKU after several months of a variety of tests. This was the first they had ever heard about this disease, and the information found on the internet was alarming. That is why when they heard that AKU patient Robert Gregory was setting up an AKU Society with his doctor Lakshminarayan Ranganath, Nick immediately went to see them in Liverpool to see what could be done.
In the early days, the AKU Society focused on providing a website with the latest information about AKU for patients around the world. Then an AKU Information Centre was set up based at the Royal Liverpool University Hospital, whose job was to contact all 50,000 GPs in the UK in order to identify patients with AKU.
Meanwhile, the research was gathering pace thanks to James Gallagher who set up a basic science programme at the University of Liverpool. The AKU Society funded a cell model of the disease, followed by an animal model and a natural history study.
In 2012, the AKU Society was instrumental in securing funding to set up the world’s first Alkaptonuria treatment centre, based at the Royal Liverpool University Hospital under Lakshminarayan Ranganath’s leadership. A key aim was to provide all the support that had been lacking until then for AKU patients, such as dietary advice, systematic screening and advice on jobs and lifestyle.
That same year, the group also secured £5 m in funding from the European Commission in order to implement a clinical development programme for a promising drug called nitisinone.
All this shows what can be achieved when a patient group works closely with clinicians and scientists in order to help patients with an ultra-rare disease.
History of AKU
AKU (OMIM 203500) is an ancient disease; there are several reports of suspected ochronosis and spondyloarthropathy in Egyptian mummies, including Harwa, which is currently housed in the Field Museum in Chicago and dates from 1500 BC. 1
Scribonius (1584) provided the earliest record of someone passing dark urine in which a school boy passed urine ‘as dark as ink’. Twenty-five years later, Schenck (1609) reported a similar phenomenon in a Carmelite monk. Similar cases were reported over the next 200 years.
The name Alkaptonuria was first introduced by Böedeker in 1859 2 who analysed urine from a 44-year-old man with lumbar spine pain and poor mobility. He identified a substance that reduced alkaline copper solutions, but the patient did not have any symptoms of diabetes, and unlike glucose, the urinary substance would not reduce alkaline bismuth. He also observed that when left to stand, the urine darkened. He further described that this occurred from the surface of the solution downwards and that this darkening accelerated on addition of alkali substances and quickly took up a large volume of oxygen. He described this new substance ‘alkapton’.
Virchow 3 later provided the first description of pigmented tissues in AKU and coined the term ‘ochronosis’ because of how homogentisic acid (HGA) pigment appeared to be ‘ochre’ under microscopy. Black pigmentation was observed in large weight-bearing joints including intervertebral discs, menisci, laryngeal and tracheal cartilage. The ligaments and synovial tissues displayed pigmentation to a lesser extent than cartilage. When the black pigmentation was observed macroscopically, it appeared yellow.
The chemical structure of ‘alkapton’ was identified in 1891, 4 and named HGA (Figure 1) because of its structural similarity to gentisic acid.
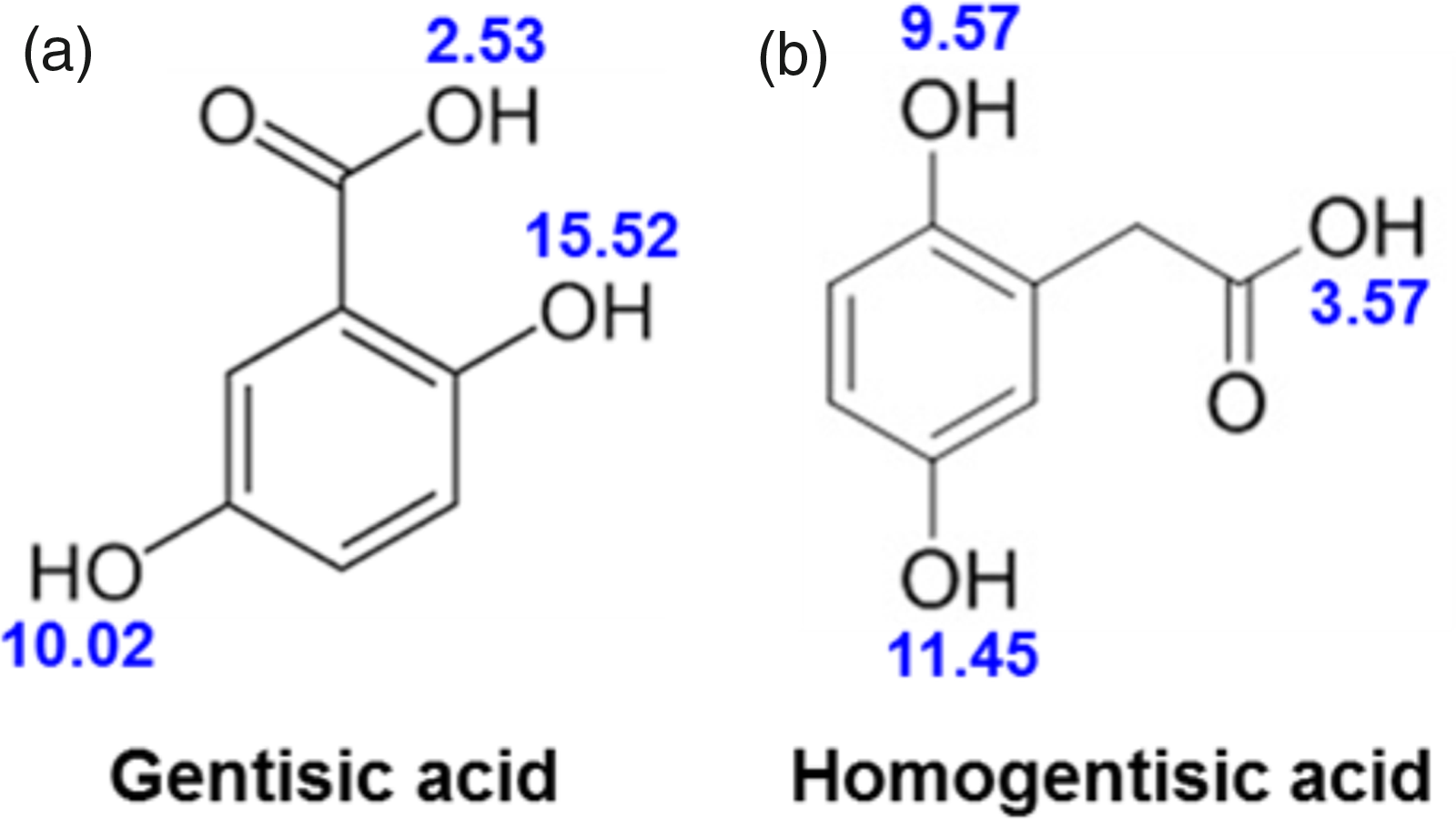
Chemical structures and associated pKa’s (denoted in blue) of (a) gentisic acid and (b) homogentisic acid.
It was in 1902 that Albrecht made the connection between ochronosis of tissues, ochronotic arthritis and AKU. Later in 1904, Osler was the first person to publish evidence to document the diagnosis of AKU in living individuals. The sufferer in question displayed pigmentation of the ears and the sclera, the latter being termed as ‘Osler’s sign’.
At this time, the disorder was described as the result of an alternative course of metabolism, harmless and usually congenital and lifelong. 5 Most famously, Garrod presented his work at the Croonian lectures in 1908, whereby he introduced the concept of ‘inborn errors of metabolism’ to demonstrate that albinism, AKU, cystinuria and pentosuria were all resultant of Mendelian inheritance, causing variation in metabolism of normal metabolites in biochemical pathways. 6
Fifty years later 7 , homogentisate 1,2-dioxygenase (HGD; E.C.1.13.11.5), an enzyme involved in the metabolism of tyrosine, was identified as the enzymatic defect responsible for AKU (Figure 2).
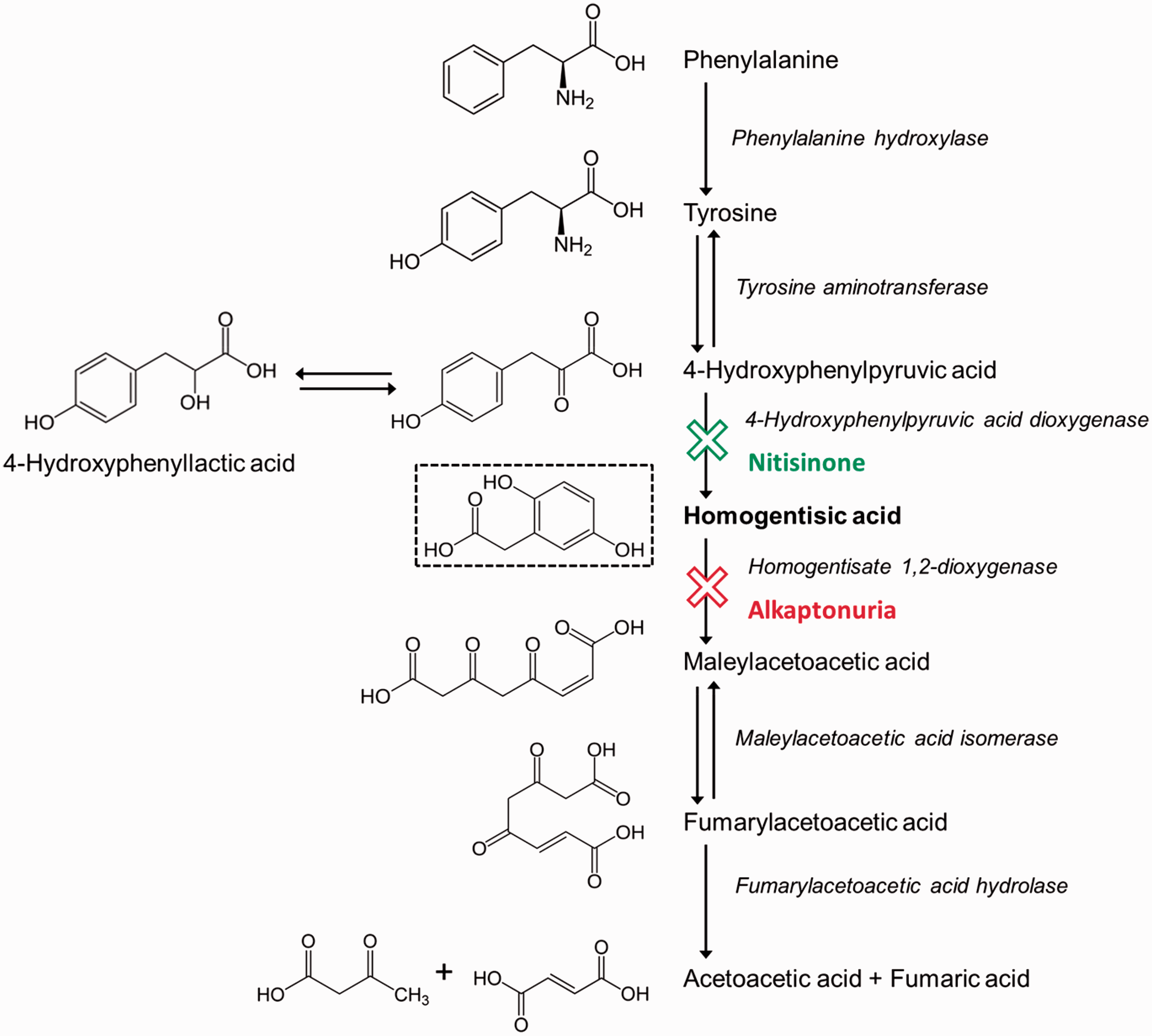
Tyrosine metabolic pathway (adapted from Taylor et al. 8 ) – highlighting the site of the enzyme defect observed in AKU and the site of action of nitisinone, a reversible competitive inhibitor of 4-hydroxyphenylpyruvic dioxygenase (HPPD EC 1.13.11.27).
Genetic basis of AKU
The HGD gene was first cloned and sequenced from Aspergillus nidulans. 9 The gene maps to the human chromosome 3q13.33 (http://www.ncbi.nlm.nih.gov/gene/3081, Gene ID: 3081) and is a single-copy gene spanning 54 363 bp of genomic sequence, with 14 exons coding for a protein of 445 amino acids that assembles into a functional hexamer arranged as a dimer of trimers. 10 Liver and kidneys are the major sites of HGD activity.
AKU arises from homozygous or compound heterozygous mutations in the HGD gene. There have been 212 unique AKU mutations identified (HGD mutation database: http://hgddatabase.cvtisr.sk, accessed 3 June 2019), of which the most frequent are missense variants (67 %), followed by splicing (12.2 %) and frameshift (12.2 %).
AKU mutations are distributed throughout the entire HGD gene with some prevalence in exons 3, 6, 7, 8 and 13. 11 Haplotype analysis has been used to trace the migration of specific AKU alleles through human history. The three most widespread AKU mutations in Europe, M368V, V300G and P230S 11 (20 %, 5 % and 5 % of European AKU mutations, respectively), appear to be ancient mutations that were introduced into Europe with the founder populations. Some HGD mutations are found in many countries, examples include S59fs which was one of the first identified AKU mutations, V300G, as well as the most frequent European mutation M368V. 11 On the other hand, there are mutations rather unique to specific regions; for example c.87 + 1G>A (p.(Tyr6_Gln29del)) for the gypsy community Narikuravar in India, 12 A122V in Jordan 13 and C120W for the Dominican Republic. 14 The largest number of cases of AKU (>200) have been reported in Slovakia, 15 the majority clustering in a small region in the north-west of the country. To date, there is no explanation for this massive increase in the incidence of AKU causing mutations in this region; it cannot be explained by a classical founder effect. In contrast, the high incidence of AKU in the Dominican Republic arose through a classical founder effect with C120W as the mutation. 14
As yet, no relationship between specific mutations and clinical manifestations has been established, although several AKU mutations have been identified, which have been shown to have residual catalytic activities in functional assays. Interestingly, for eight AKU patients, no HGD mutations were identified, and in 22 cases, only one mutant allele was found. 13
Recently, a large genomic deletion of exon 2 including intronic sequences was reported in one case from Lebanon 16 and has also been identified in two siblings from Israel. 13 It is possible that, in addition to deep intronic mutations, large deletions encompassing one or more exons might be occurring in cases where genomic sequencing does not lead to mutation identification.
Animal models
In 1994, an AKU mouse model generated by ethylnitrosourea (ENU) mutagenesis was identified by Montagutelli et al. 17 due to the presence of darkened cage bedding caused by elevated HGA in the urine. The AKU mutation was backcrossed onto the BALB/cByJ and C57BL/6J murine backgrounds. The murine HGD gene was cloned and the mutation was identified to be a splice mutation that results in a truncated HGD protein. 18 This mouse harbours a recessive splice site mutation, c.1006 + 2T > A, in exon 10 of HGD gene (GenBank NM_013547) located on chromosome 16. This mutation leads to skipping of one or two exons and the generation of a premature stop codon. Initially, it was reported that the AKU mouse model had the metabolic defects of AKU, but that the tissues did not undergo ochronosis. Several theories were suggested including the short life span of the mouse and the endogenous production of ascorbic acid. However, it was subsequently demonstrated that this AKU model (BALB/c HGD–/–) has relatively stable elevated plasma HGA concentrations and extensive chondrocyte pigmentation, via a modified Schmorl’s stain. 19 This model showed that initial pigmentation early in life is pericellular, progressing linearly with age to the intracellular compartment. Treatment with a drug called nitisinone, which blocks the enzyme HPPD forming HGA, was shown to completely prevent chondrocyte pigmentation in mice.20,21
This HGD–/– AKU mouse model has greatly contributed to our knowledge of AKU, in terms of both the disease pathology and treatment; however, ENU mutagenesis is not a targeted approach to create a model of genetic disease. Due to the high frequency of DNA mutations that ENU causes, there could potentially be other unknown and uncharacterized mutations in this mutagenesis model affecting the disease phenotype. To overcome this uncertainty, a new targeted mouse model of AKU has been raised in the C57BL/6 background using a mutant knockout-first allele obtained from the KOMP repository (www.komp.org). In addition to being targeted, the knockout-first model harbours a lacZ/lacZ transgene for localizing gene expression and can be manipulated via FLP/FRT and Cre/LoxP recombination to obtain an inducible and tissue-specific knockout. The conditional allele can be used to investigate the effect of partial HGD knockout on the AKU phenotype. With gene therapy becoming a likely tool in the future, this conditional mouse model provides an insight into the level and location of HGD expression that could rescue the AKU phenotype in humans.
Clinical manifestations observed in patients
Helliwell et al. 22 demonstrated at autopsy the plethora of features observed in AKU confirming the true multisystem nature of the disease (Figure 3). There are few signs and symptoms in the young 23 with symptoms appearing around age 30 years and progressively increasing in severity with age, apart from dark urine and renal stones.24–26 The various clinical features in AKU can be appreciated from the Alkaptonuria Severity Score Index (AKUSSI) (Table 1). This incorporates multiple, clinically meaningful outcomes that can be described in a single score, including kidney and prostate stones, aortic stenosis, bone fractures, tendon/ligament/muscle ruptures, kyphosis, scoliosis, joint replacements and all other clinical features of AKU.
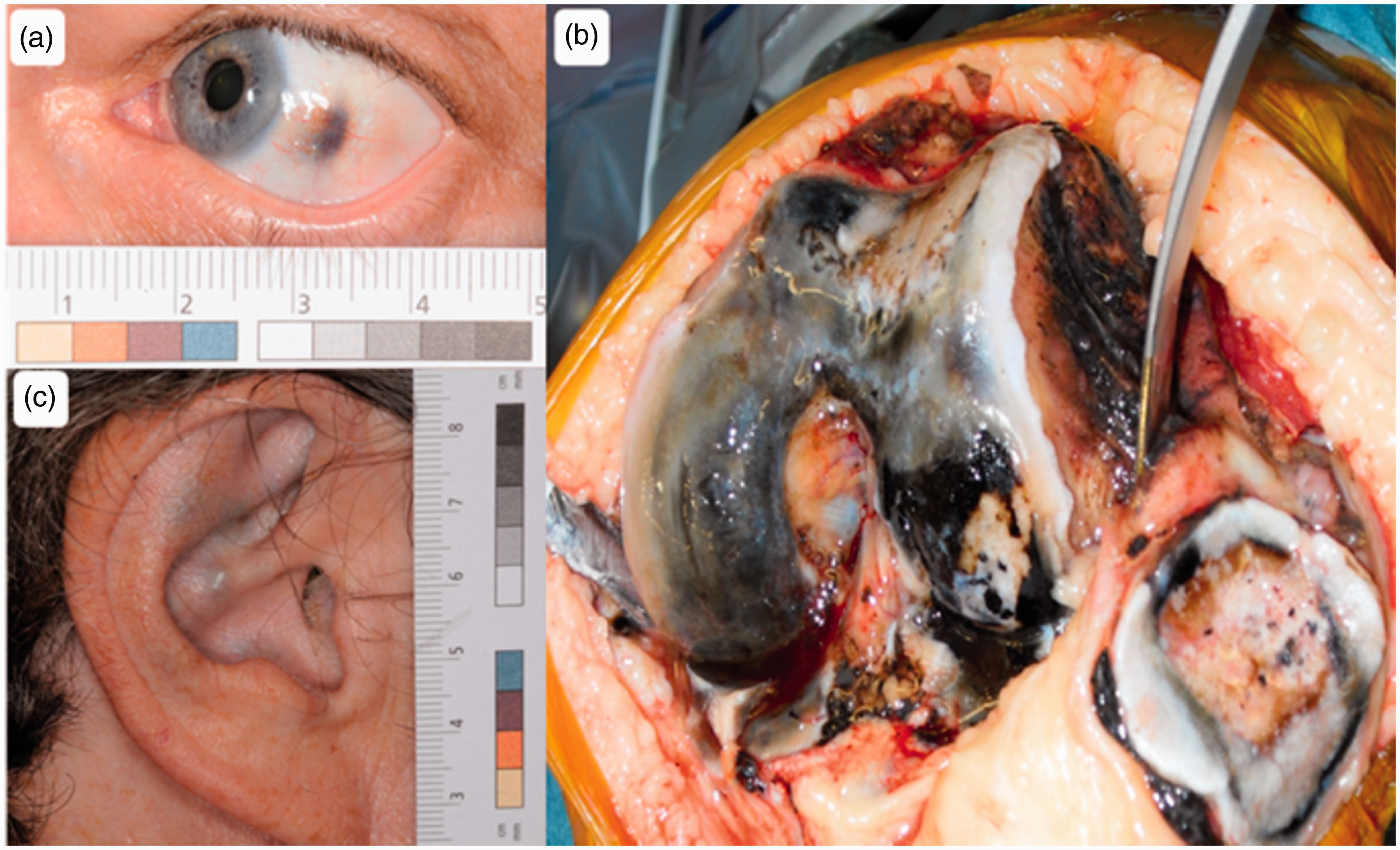
Clinical features frequently observed in AKU that should alert a clinician to its possible diagnosis. (a) Pigmentation of the sclera of the eye, (b) extensive osteoarthritis of the large joints. Here is an image of a knee joint at surgery showing pigmentation and fragmentation of the articular cartilage and (c) slate grey pigmentation of cartilage in the ear.
Summary of Alkaptonuria Severity Score Index (AKUSSI).
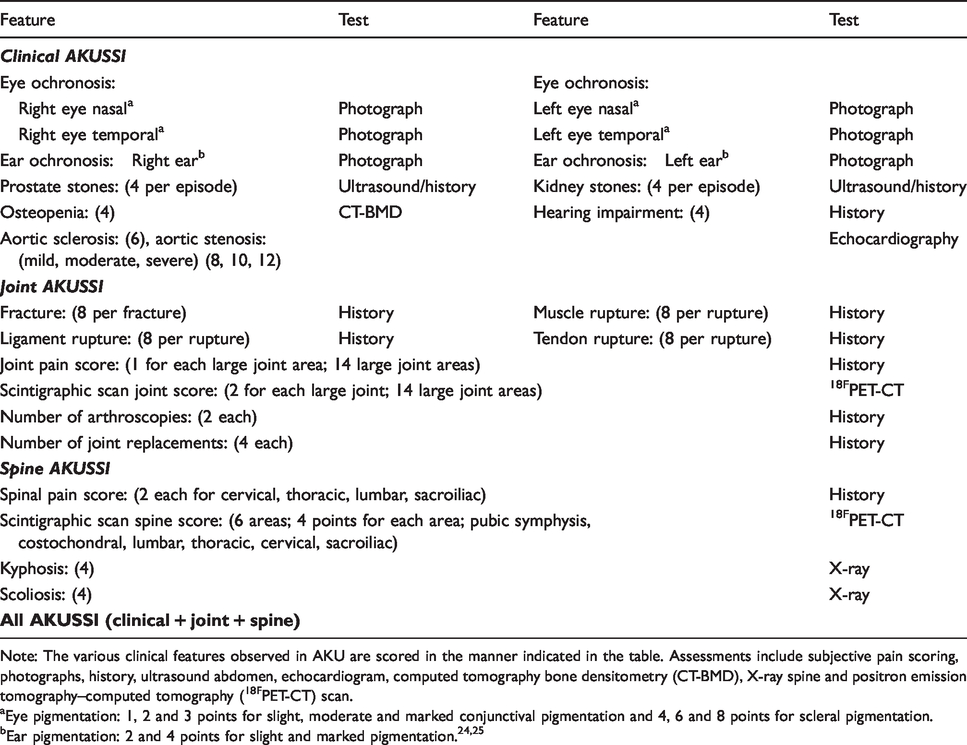
Note: The various clinical features observed in AKU are scored in the manner indicated in the table. Assessments include subjective pain scoring, photographs, history, ultrasound abdomen, echocardiogram, computed tomography bone densitometry (CT-BMD), X-ray spine and positron emission tomography–computed tomography (18FPET-CT) scan.
Eye pigmentation: 1, 2 and 3 points for slight, moderate and marked conjunctival pigmentation and 4, 6 and 8 points for scleral pigmentation.
Biochemical consequences
AKU is a biochemical defect that arises in the tyrosine degradation pathway (Figure 2), whereby deficiency of HGD results in significantly elevated serum and urine HGA.7,27–30 With advancements in analytical methodologies, HGA has been quantified to be present in millimolar concentrations in urine from AKU patients27–30; this is in contrast to healthy individuals, where urinary HGA has been reported as <1.1 μmol/L. 31 Our group reported the circulating concentration of HGA using liquid chromatography tandem mass spectrometry (LC-MS/MS) and although an order of magnitude lower than excreted concentrations, HGA circulates in micromolar quantities in AKU.29,30 Of note, although AKU is a defect in the tyrosine degradation pathway, serum and urine tyrosine are within the reference range prior to administration of nitisinone.27–30,32
Management and treatment
There is still no approved disease-modifying therapy for AKU. Gene and enzyme replacement therapies are still some way off, and currently, treatment remains supportive and palliative (Table 2). Low protein diet has been used with little effect,27,33 and ascorbic acid used as an antioxidant to prevent the conversion of HGA to ochronotic pigment has unproven efficacy. 27 Lifestyle counselling and physiotherapy can be beneficial to patients but are underutilized.
Supportive and palliative treatment modalities available for AKU.
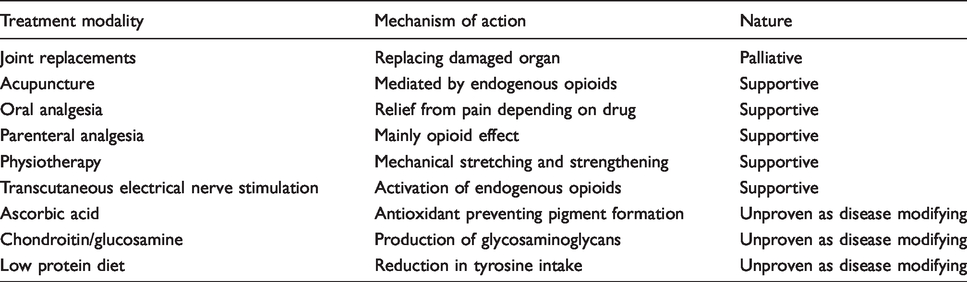
Supportive analgesia and palliative arthroplasty are crucial to managing the severe musculoskeletal pain in AKU.26,27,34,35 Local anaesthetic patches such as lidocaine are also beneficial. Intermittent colchicine has also been used to control episodic pain. 34 Transcutaneous electrical nerve stimulation and acupuncture are widely used to control pain in patients attending the National AKU Centre (NAC). Neuromuscular blocks and trigger point injections can also provide long-term analgesia in AKU. Joint replacement is inevitable and highly effective. Spinal decompression surgery is needed when spinal compression complicates AKU.35–37 Aortic valve disease is highly prevalent and is almost universal by age 60 years in AKU. 38 Aortic valve surgery is better carried out electively before left ventricular decompensation ensues, but is technically challenging in ochronotic aortic tissue and sometimes fatal. Stone disease is also common in AKU due to increased HGA. To minimize the formation of renal stones and prevent renal impairment, it is important to emphasize good hydration.
Disease-modifying therapy through inhibition of 4-HPPD
Nitisinone (2-[2-Nitro-4-(trifluoromethyl)benzoyl]-1,3-cyclohexanedione, C14H10F3NO5, MW 329.2) is a reversible competitive inhibitor of 4-HPPD in the tyrosine metabolic pathway (Figure 2). 39 Developed as a herbicide, nitisinone was found to be highly efficacious in hereditary tyrosinemia type 1 (HT1, OMIM 276700), which is now standard first-line therapy.39–41 The mode of action of nitisinone led to the recognition that it could be effective in AKU. Decreasing circulating concentrations of HGA through the inhibition of 4-HPPD should in principle decrease ochronosis, thus preventing the progression of disease in AKU. As nitisinone targets and corrects the main metabolic abnormality responsible for AKU, it should prevent or reduce its associated morbidity, if started before the symptomatic phase, and alter progression in those already symptomatic. There are several key studies which have looked at the key metabolites in the tyrosine pathway, both before and after a trial of nitisinone therapy (Table 3).
Summary of clinical studies that have evaluated nitisinone for the treatment of AKU.
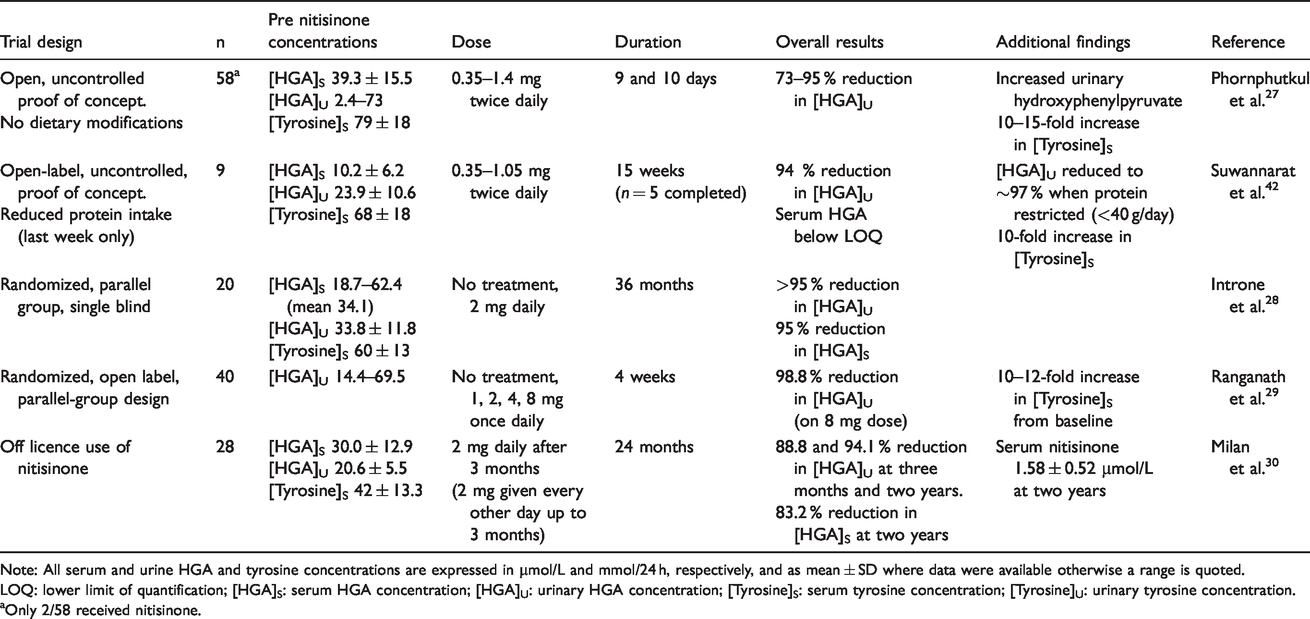
Note: All serum and urine HGA and tyrosine concentrations are expressed in µmol/L and mmol/24 h, respectively, and as mean ± SD where data were available otherwise a range is quoted.
LOQ: lower limit of quantification; [HGA]S: serum HGA concentration; [HGA]U: urinary HGA concentration; [Tyrosine]S: serum tyrosine concentration; [Tyrosine]U: urinary tyrosine concentration.
Only 2/58 received nitisinone.
Clinical trials of nitisinone at the National Institute of Health, USA
In the earliest study of AKU 27 (n = 58), two women were given nitisinone at a 30-fold lower dose than that used in HT1, 41 urine and plasma HGA, and serum tyrosine concentrations were measured in all patients pre- and post-treatment. Within this study, there were also 10 patients on ascorbic acid doses ranging from 0.25 to 4 g/day (recommended daily dose is 0.065–0.09 g/day, with an upper limit of 2 g). Notable in this study was that there was no correlation with urine HGA excretion and genetic mutation. 27 High-dose ascorbic acid treatment has not been proven efficacious.27,43,44 The mean urinary HGA in those on high-dose ascorbic acid was not significantly different from the untreated AKU patients. In addition, those on a low protein diet also showed no difference in urinary HGA excretion.
Another open-label study at the National Institute of Health (NIH) employed nitisinone (2.1 mg daily), demonstrating a 95 % decrease in urinary HGA excretion in nine AKU adult patients over a four-month period. 42 This study also confirmed that a strict protein restriction (one week only) produced an additional decrease in urine HGA concentrations (Table 3). Various definitions of protein restriction led to some of the ambiguity in effects with the recommended daily allowance (RDA) defined as 0.8 g protein/kg of body weight; approximately 56 or 46 g/day for the average sedentary male or female, respectively. Where a reduction in urinary HGA has been quantitated, protein has been restricted to <40 g/day and as low as 20 g/day, 42 which is not sustainable long-term and would lead to detrimental effects on muscle strength, a side-effect which would worsen these patients’ symptoms.
In a third study, a three-year single-blind clinical trial of 20 patients, the nitisinone group (2 mg/day) showed a sustained decrease in mean urinary HGA from 30.3 to 0.74 mmol/day. 28 Mean plasma HGA concentrations fell from 34.1 to 1.82 μmol/L after treatment. Urine and plasma HGA decreased by 98 and 95 %, respectively, but despite this, the study reported inconclusive, as the clinical primary and secondary outcome measures did not show benefit from the treatment.
Clinical trials of nitisinone in the UK and Europe
The impetus for continued clinical development of nitisinone use in AKU came from Liverpool in the UK. A two-pronged strategy was adopted: (1) licensing of nitisinone for AKU through clinical trials and (2) use of nitisinone off-label to determine its efficacy and safety in AKU. The first clinical trial was a dose-ranging study (part of a programme called DevelopAKUre, funded by the European Commission as part of the Framework Programme 7) called SONIA 1 (Suitability Of Nitisinone In Alkaptonuria 1) and concluded that nitisinone 8 mg was the dose that decreased urine HGA most efficaciously. 29 Nitisinone 10 mg capsule once daily is now being trialled in SONIA 2 (Suitability Of Nitisinone In Alkaptonuria 2); this is a four-year outcomes study where the clinical phase is due to be completed in 2019. If successful, it should enable an application to the European Medicines Agency to approve the use of nitisinone in AKU.
Off-label use of nitisinone
In 2012, NHS England Highly Specialised Services designated Liverpool as the NAC and approved the use of nitisinone 2 mg daily. This off-label use of nitisinone in AKU is carried out by multidisciplinary team of health-care professionals. While the nitisinone is being prescribed as part of providing a service to patients, as required by NHS England, the service is ‘protocolized’ to allow collection of high-quality data to determine the efficacy and safety of nitisinone. The three-year data on the use of nitisinone in the NAC show partial reversal of ochronosis and slower progression of the disease.45,46 This is the first time that the disease process and outcomes in AKU have been beneficially modified by any therapy.
Nitisinone-induced hypertyrosinaemia
The universal metabolic complication of nitisinone therapy is hypertyrosinaemia, reported in both HT140,47,48 and AKU.28–30,32,42 The dose of nitisinone used in AKU trials is significantly lower compared with that in HT1; mg/day compared with 1–2 mg/kg body weight in HT1; however, the increase in tyrosine is comparable. Clinically, in HT1, treatment is commenced from an early age and with the knowledge of cognitive impairment,49–51 serum tyrosine concentrations are regularly monitored and as a guide the aim should be to keep tyrosine concentrations between 200 and 400 μmol/L up to the age of about 12 years. This is not easy to achieve, and some centres allow plasma tyrosine concentrations up to 500 μmol/L. 52
There is no direct evidence of altered cognition or neurotransmitter metabolism in AKU patients with hypertyrosinaemia. A short-term study has demonstrated increased urinary excretion of the dopamine metabolite, 3-methoxytyramine (3-MT) and decreased urinary normetadrenaline over a four-week period. 53 The increased urinary 3MT was confirmed in patients attending the NAC over a two-year period with concentrations increasing two-fold of the reference range. 54 The marked increase in 3MT excretion suggests that nitisinone alters peripheral metabolism of catecholamines, more specifically dopamine. Metabolism of catecholamine neurotransmitters is complex due to the multiple origins, and urine concentrations will reflect circulating concentrations, renal uptake and renal synthesis. 55 Of interest in this larger cohort, there was no concurrent change in Beck’s Depression Inventory II scores associated with these patients supporting the renal synthesis hypothesis. 54 Interestingly, the previous change in normetadrenaline was not reflected over this longer time period; with the increased patient numbers and monitoring of patients over two years, the NAC data are likely to reflect changes in those on long-term therapy. Reassuringly, a study using mass spectrometry imaging demonstrated that monoamine neurotransmitter patterns in brain tissue from a murine model of AKU did not change following treatment with nitisinone. 56
Despite careful use of nitisinone, tyrosine ocular keratopathy, as well as skin rash are observed in approximately 5 % treated with the 2 mg dose.28,29,57–59 With circulating tyrosine concentrations around >800 μmol/L, the solubility of tyrosine is exceeded and corresponds to ocular tyrosine concentrations of 3500 μmol/L, 60 the point at which tyrosine crystallizes in the cornea leading to corneal keratopathy; similar critical tyrosine thresholds for cutaneous and brain effects have not been described. Published cases have demonstrated that eye symptoms resolve upon cessation of nitisinone and either a lower dose and or protein restriction enables the patient to re-start nitisinone. In HT1, studies have also demonstrated ocular keratopathy (∼9 %) in patients on nitisinone treatment.61,62
Introne et al. 28 demonstrated that the average serum tyrosine was approximately 800 μmol/L with the highest measured being 1500 μmol/L on a 2 mg daily dose (no dietary restriction). Ranganath et al. 29 added to this with SONIA 1 trial, whereby a short-term study examined the effects of varying nitisinone doses (1–8 mg daily) on the metabolic profile. A dose-dependent decrease in urine HGA excretion was measured with a 98.8 % reduction at 8 mg/day, which was not reflected in serum tyrosine concentrations; although there was the expected 10-fold increase, this was not dose dependent. This supports the findings of Introne et al. 28 with all patients having serum tyrosine >500 μmol/L and the highest 1117 μmol/L (on 4 mg daily dose). Data from the NAC corroborate these previous studies with the longitudinal monitoring of tyrosine metabolites while treated with a 2 mg daily dose of nitisinone with a 94 % reduction in urine HGA maintained at two years with a concurrent serum HGA reduction of 83.2 %. 30 Mean serum tyrosine at two years was 594 μmol/L with a large variation reflecting the dietary protein contribution.
Rationale for the treatment of nitisinone-induced hypertyrosinaemia
Limiting phenylalanine and tyrosine amino acid intake should mitigate hypertyrosinaemia. The minimum dietary recommendations for optimal health are phenylalanine and tyrosine intakes of 15 mg/kg/day, with an optimal dietary ratio of phenylalanine and tyrosine in mass units of 60:40, similar to the phenylalanine to tyrosine ratio seen in human tissue. 63
Tackling hypertyrosinaemia by switching to elemental amino acid administration free of phenylalanine and tyrosine is impractical. Instead, the emphasis is on decreasing dietary protein intake to manage hypertyrosinaemia. It is necessary to ensure that the reference nutrient intake (RNI) for protein intake is met, the RNI for protein is 0.75 g/kg of body weight in the UK 64 and the RDA for protein is 0.8 g/kg of body weight in the USA.65,66 Meeting these targets for protein should ensure that the requirement for limiting amino acids such as lysine, methionine, threonine and tryptophan are met; such an approach will guarantee adequate provision of other amino acids including phenylalanine and tyrosine.
There is a lack of guidance in the management of hypertyrosinaemia in adults, although there is some guidance in children with HT1.52,63 The goal in these children is to maintain serum tyrosine between 200 and 400 μmol/L and to avoid concentrations >500 μmol/L. 52 Such stringent goals are difficult to achieve in adults with AKU as they have been used to consuming a relatively normal protein intake. At the NAC, a pragmatic algorithm-based approach is employed, consistent with existing knowledge and recommendations, where the goal is to maintain circulating tyrosine as low as possible <900 μmol/L. Values of circulating tyrosine of <500 μmol/L are considered desirable, and protein intake of 1 g/kg is considered acceptable. Action thresholds have been devised between 500 and 700, and between 700 and 900 μmol/L, invoking lower protein intakes of 0.9 and 0.8 g/kg body weight, respectively,29,45 while meeting minimum daily requirements. Concentrations >900 μmol/L require, in addition, phenylalanine/tyrosine-free meal exchanges.
Timing nitisinone therapy and the natural history of AKU
AKU, present from birth, appears to have few features in the young. Although it was believed that ocular and ear pigment only appeared around age 30 years, careful assessment by direct tissue studies and ocular photographs has shown the presence of ocular pigment even at age 16 years (unpublished, personal communication with Ranganath LR). A previous report in Slovakia describes the presence of ear and scleral pigment at age 12 and 13 years, respectively, but no images are presented. 23
Analytical methods for measurement of nitisinone, tyrosine, HGA
Key to the studies described within this review are the analytical methodologies employed for the quantification of HGA and tyrosine-related metabolites that have been measured in patients with AKU (Table 4). Many of these methods involve sample preparation coupled with an extensive and often complex analytical technique, rendering many of these methods impractical in a modern busy clinical laboratory.
Summary of targeted analytical methodologies reported for the measurement of HGA, tyrosine and nitisinone in patients with AKU.
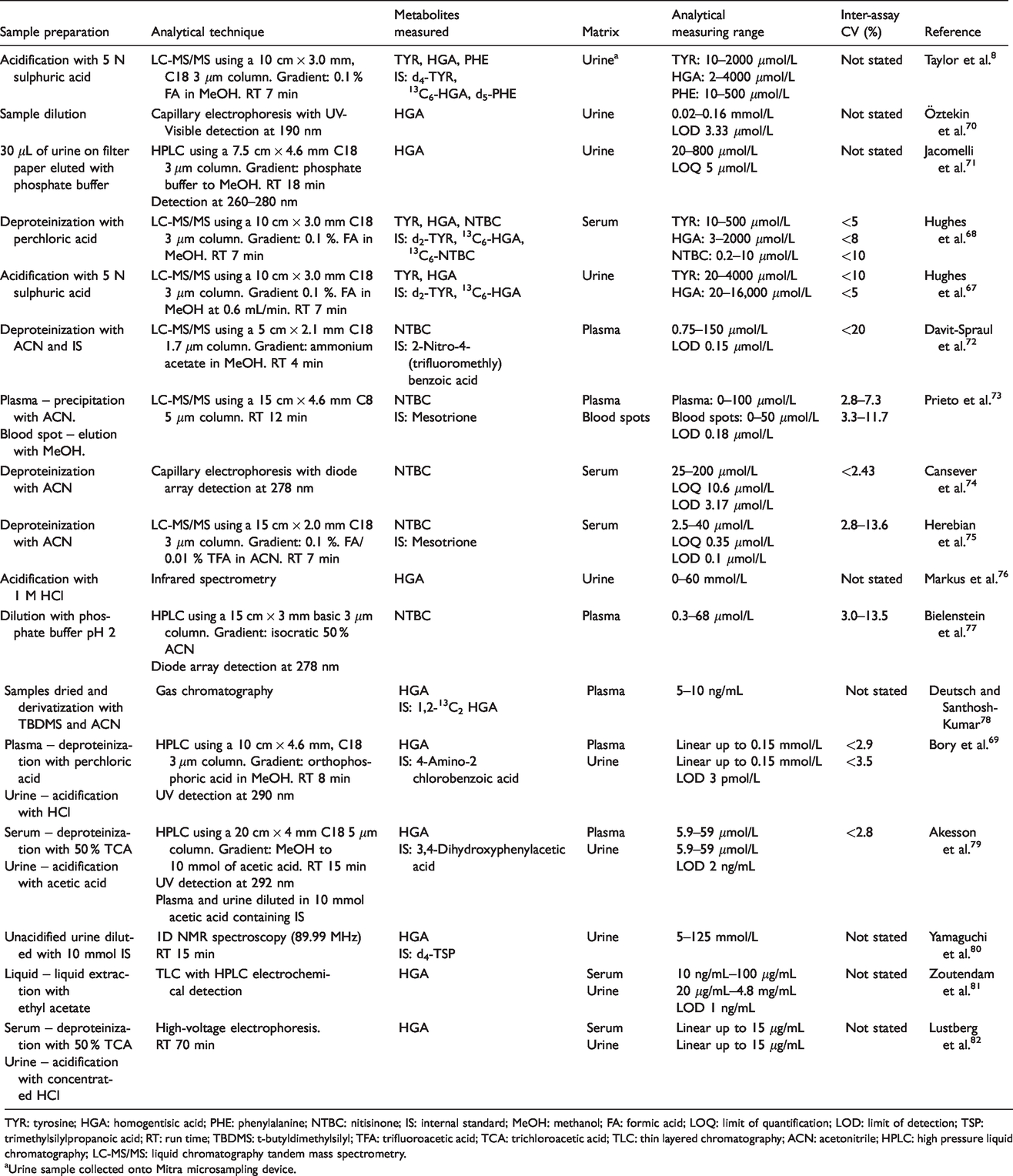
TYR: tyrosine; HGA: homogentisic acid; PHE: phenylalanine; NTBC: nitisinone; IS: internal standard; MeOH: methanol; FA: formic acid; LOQ: limit of quantification; LOD: limit of detection; TSP: trimethylsilylpropanoic acid; RT: run time; TBDMS: t-butyldimethylsilyl; TFA: trifluoroacetic acid; TCA: trichloroacetic acid; TLC: thin layered chromatography; ACN: acetonitrile; HPLC: high pressure liquid chromatography; LC-MS/MS: liquid chromatography tandem mass spectrometry.
Urine sample collected onto Mitra microsampling device.
The analytical platform adopted for measurement of tyrosine and related metabolites at the NAC is LC-MS/MS.67,68 Use of this analytical methodology has allowed for the validation of quantitative methods for serum and urinary metabolites. An important preanalytical consideration is that samples must be acidified prior to analysis to minimize the degradation of HGA and tyrosine-related metabolites. 69
Metabolomics
Traditionally, the diagnosis of inborn errors of metabolism have heavily relied on targeted analysis in biological fluids, including urine, serum and cerebrospinal fluid using a range of analytical techniques including chromatography and mass spectrometry-based techniques. Over the last 10 years, there has been increasing interest in utilizing a non-targeted ‘hypothesis-free’ approach to evaluate the metabolome. 83
Typically, high-resolution accurate mass spectrometry and nuclear magnetic resonance techniques are used when adopting this approach; this offers major potential benefits for improving our understanding of metabolic disease and response to treatment, and the delivery of personalized healthcare. 84 The major challenges in performing non-targeted metabolomics in a clinical setting are study design, 85 quality control 86 and the validation of analytical and chemometric methodologies.87–89
Gertsman et al. 90 was the first to report on the use of high-resolution accurate mass spectrometry (HRAMS) to evaluate the serum metabolome of patients with AKU taking nitisinone (2–8 mg daily) over a period of 6 months to 3.5 years. This small study revealed the expected decrease in HGA and increase in tyrosine following treatment with nitisinone. This untargeted analysis also revealed significant increases in acetyl- and γ-glutamyltyrosine, which is not surprising due to the significant hypertyrosinaemia that is observed following treatment with nitisinone. In a separate publication related to the same study, 91 they reported novel disturbances in tryptophan metabolism following treatment with nitisinone. Specifically, indole-carboxyaldehyde, indole-3-pyruvate and indole-3-lactate were shown to increase five-fold. It was proposed that the tyrosine metabolite 4-hydroxyphenylpyruvate, which increases significantly following nitisinone therapy, upregulates tryptophan aminotransferase activity resulting in downstream changes in the concentration of tryptophan metabolites and not tryptophan itself. The significance of this is unknown; it has been suggested that aromatic ketoacids increase the affinity of tryptophan transaminase for tryptophan, 92 thus altering its metabolism.
Davison et al. 93 also reported increases in serum acetyl- and γ-glutamyltyrosine following treatment with nitisinone using HRAMS, but interesting only observed a change in indole-3-lactate. This study did show increases in other tryptophan metabolites including trigonelline and quinoline carboxylic acid. Novel changes were also observed in metabolites relating to the citric acid cycle (decrease in succinate and α-ketoglutarate) and xanthine metabolism (decrease in uridine and inosine). It was suggested that these changes along with those observed in acetyl- and γ-glutamyltyrosine relate to changes in the redox state of the cell following treatment with the HGA-lowering agent nitisinone.
Increases in 4-hydroxyphenylacetate, 4-hydroxybenzaldehyde and benzaldehyde were also reported following nitisinone treatment. The significance of the latter two metabolites is uncertain but was suggested that they relate to the ochronotic pigment observed in AKU. The increase in 4-hydroxyphenylacetate is thought to result from increased activity of gut microbiota following treatment with nitisinone due to the presence of less oxidative stress.
Norman et al. 94 have also reported novel changes in urinary tryptophan and related metabolites in patients with AKU treated with nitisinone. In this study, urinary indoxyl sulphate, tryptophan and kynurenine decreased while xanthurenic acid increased following treatment. This study supports the changes reported in 4-hydroxybenzaldehyde and 4-hydroxyphenylacetate 93 but also reported novel changes in 4-coumarate, tyramine, mandelic acid and phenylacetic acid. Interestingly, a number of changes were also observed in purine metabolism. Importantly, many of these changes reported in patients were also observed in a murine model of AKU treated with nitisinone that was used in this study reinforcing that the changes observed were a consequence of nitisinone therapy.
What AKU has told us about osteoarthritis
The study of tissue from patients and murine models with AKU has significantly advanced our understanding of the pathophysiology underlying the progression of ochronosis and the related osteoarthropathy, revealing vital information on the microanatomical, cellular and biochemical changes observed in joints. It has also provided a unique platform to allow us to better understand the pathophysiology of more common diseases like osteoarthritis (OA), and has revealed significant overlap between the two diseases.
In AKU, joint destruction is related to the deposition of pigmented polymers and in OA this results from the deposition of carbohydrates. The former leading to the progression of ochronosis and the latter the formation of advanced glycation end (AGE) products. A consequence observed in both diseases is that changes to the composition and organization of the extracellular matrix, including loss of proteoglycans and disruption of collagen fibrils, are observed. The lack of protective proteoglycans means collagen is exposed and susceptible to the action of reactive pigmented polymers and AGE products, respectively. The consequence of chemical modification in OA and AKU is that collagen becomes stiff and less tolerant to mechanical loading leading to structural damage. Certainly in AKU and theoretically in OA, this process starts in calcified tissue and spreads throughout the hyaline cartilage to the articular surface. 95 This leads to abnormal transmission of mechanical loading through cartilage to underlying bone, which results in damage to the subchondral plate and load-induced remodelling, and the formation of high-density mineralized protrusions. 96 The latter was first observed in the cartilage associated with the femoral head from a patient with osteoarthropathy of AKU and subsequently found that these are also present in hip and knee joints from patients with OA. Additionally, focal loss and sclerosis of the subchondral plate, 95 as well as aberrant remodelling of the underlying bone, including formation of trabecular excrescences have been observed. 97
Conclusions
AKU is a rare, yet iconic inborn error of metabolism that was first reported to follow the laws of Mendelian inheritance by Sir Archibald Garrod over 120 years ago. Since this time, remarkable progress through collaborative working between patients, scientists and clinicians in a relatively short period of time has resulted in the use of a disease-modifying therapy for the treatment of this disease. AKU should serve as a model for other less well-understood diseases, for which there is currently no treatment. Our attempts to better understand AKU has also enabled a better understanding of the common disease OA.
Footnotes
Acknowledgements
This article was prepared at the invitation of the Clinical Sciences Reviews Committee of the Association for Clinical Biochemistry and Laboratory Medicine.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Andrew S Davison is funded through a National Institute for Health Research (NIHR) Doctoral Research Fellowship (grant code: HCS DRF-2014–05-009). This article presents independent research funded by NIHR. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care.
Ethical approval
Not applicable.
Guarantor
ASD.
Contributorship
All authors contributed to writing this article. ASD integrated contributions, revised and edited entire review.